

A unified framework for inferring the multi-scale organization of chromatin domains from Hi-C
- PMID: 33724986
- PMCID: PMC7997044
- DOI: 10.1371/journal.pcbi.1008834
A unified framework for inferring the multi-scale organization of chromatin domains from Hi-C
Abstract
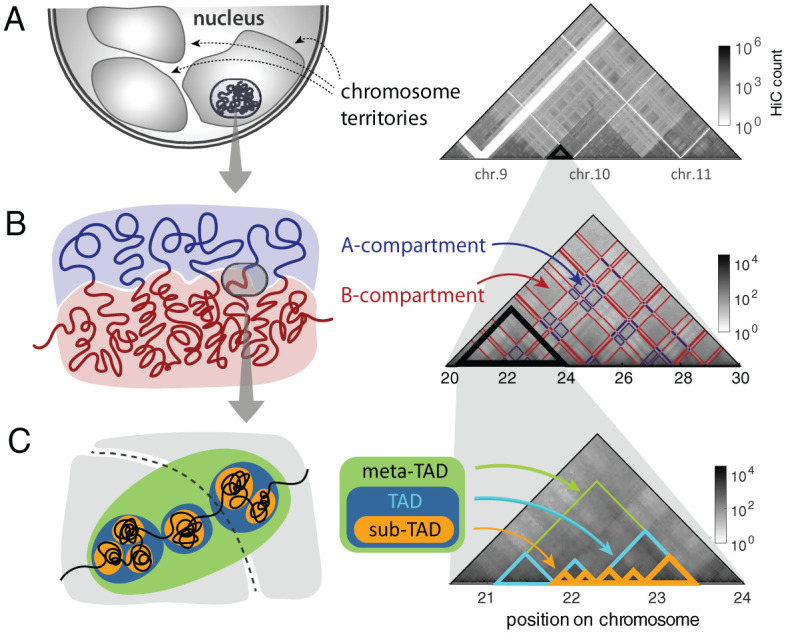
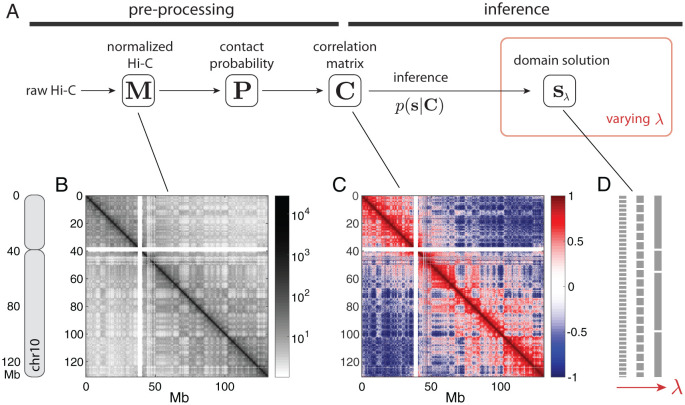
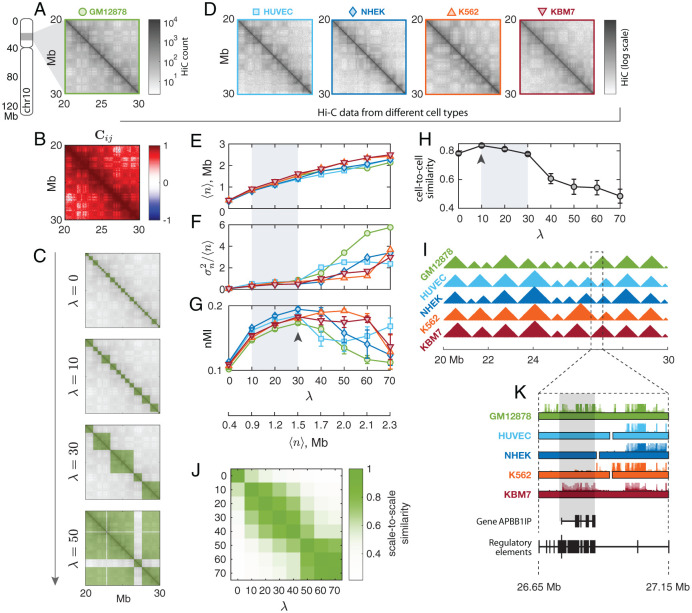
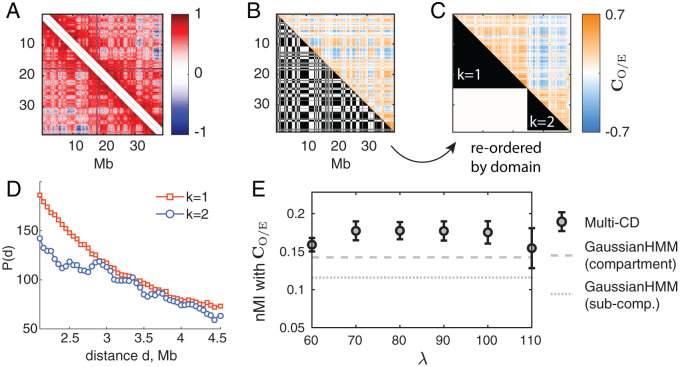
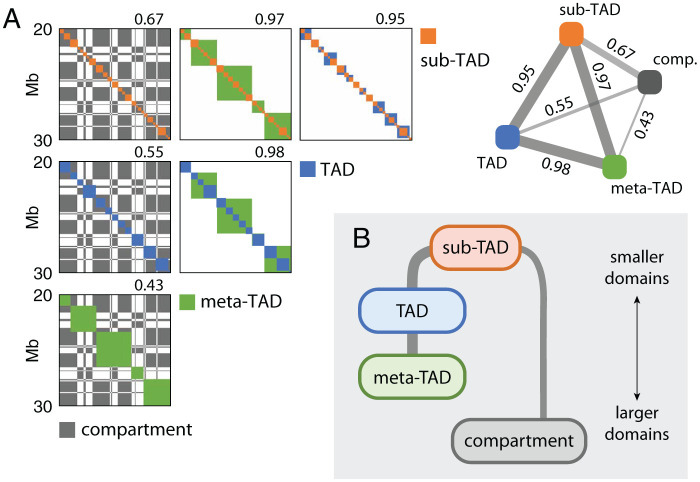
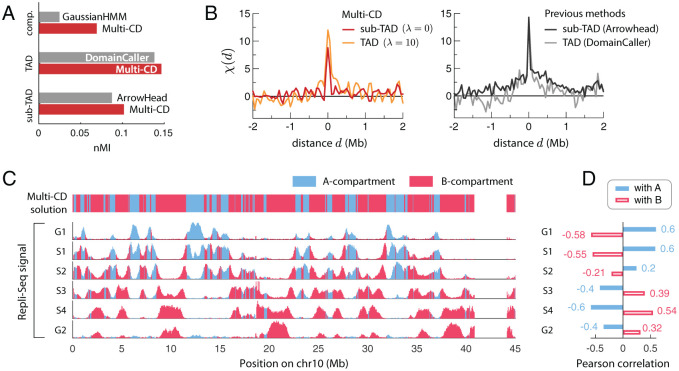
Chromosomes are giant chain molecules organized into an ensemble of three-dimensional structures characterized with its genomic state and the corresponding biological functions. Despite the strong cell-to-cell heterogeneity, the cell-type specific pattern demonstrated in high-throughput chromosome conformation capture (Hi-C) data hints at a valuable link between structure and function, which makes inference of chromatin domains (CDs) from the pattern of Hi-C a central problem in genome research. Here we present a unified method for analyzing Hi-C data to determine spatial organization of CDs over multiple genomic scales. By applying statistical physics-based clustering analysis to a polymer physics model of the chromosome, our method identifies the CDs that best represent the global pattern of correlation manifested in Hi-C. The multi-scale intra-chromosomal structures compared across different cell types uncover the principles underlying the multi-scale organization of chromatin chain: (i) Sub-TADs, TADs, and meta-TADs constitute a robust hierarchical structure. (ii) The assemblies of compartments and TAD-based domains are governed by different organizational principles. (iii) Sub-TADs are the common building blocks of chromosome architecture. Our physically principled interpretation and analysis of Hi-C not only offer an accurate and quantitative view of multi-scale chromatin organization but also help decipher its connections with genome function.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
Similar articles
-
MrTADFinder: A network modularity based approach to identify topologically associating domains in multiple resolutions.PLoS Comput Biol. 2017 Jul 24;13(7):e1005647. doi: 10.1371/journal.pcbi.1005647. eCollection 2017 Jul. PLoS Comput Biol. 2017. PMID: 28742097 Free PMC article.
-
Reciprocal insulation analysis of Hi-C data shows that TADs represent a functionally but not structurally privileged scale in the hierarchical folding of chromosomes.Genome Res. 2017 Mar;27(3):479-490. doi: 10.1101/gr.212803.116. Epub 2017 Jan 5. Genome Res. 2017. PMID: 28057745 Free PMC article.
-
Methods for the Analysis of Topologically Associating Domains (TADs).Methods Mol Biol. 2022;2301:39-59. doi: 10.1007/978-1-0716-1390-0_3. Methods Mol Biol. 2022. PMID: 34415530
-
The 3D Genome: From Structure to Function.Int J Mol Sci. 2021 Oct 27;22(21):11585. doi: 10.3390/ijms222111585. Int J Mol Sci. 2021. PMID: 34769016 Free PMC article. Review.
-
A (3D-Nuclear) Space Odyssey: Making Sense of Hi-C Maps.Genes (Basel). 2019 May 29;10(6):415. doi: 10.3390/genes10060415. Genes (Basel). 2019. PMID: 31146487 Free PMC article. Review.
Cited by
-
The shape of chromatin: insights from computational recognition of geometric patterns in Hi-C data.Brief Bioinform. 2023 Sep 20;24(5):bbad302. doi: 10.1093/bib/bbad302. Brief Bioinform. 2023. PMID: 37646128 Free PMC article. Review.
-
Mapping the semi-nested community structure of 3D chromosome contact networks.PLoS Comput Biol. 2023 Jul 11;19(7):e1011185. doi: 10.1371/journal.pcbi.1011185. eCollection 2023 Jul. PLoS Comput Biol. 2023. PMID: 37432974 Free PMC article.
-
Structural basis for the preservation of a subset of topologically associating domains in interphase chromosomes upon cohesin depletion.Elife. 2024 Mar 19;12:RP88564. doi: 10.7554/eLife.88564. Elife. 2024. PMID: 38502563 Free PMC article.
-
ENT3C: an entropy-based similarity measure for Hi-C and micro-C derived contact matrices.NAR Genom Bioinform. 2024 Jul 2;6(3):lqae076. doi: 10.1093/nargab/lqae076. eCollection 2024 Sep. NAR Genom Bioinform. 2024. PMID: 38962256 Free PMC article.
-
A comprehensive benchmarking with interpretation and operational guidance for the hierarchy of topologically associating domains.Nat Commun. 2024 May 23;15(1):4376. doi: 10.1038/s41467-024-48593-7. Nat Commun. 2024. PMID: 38782890 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
