

Large-Scale Metagenome Assembly Reveals Novel Animal-Associated Microbial Genomes, Biosynthetic Gene Clusters, and Other Genetic Diversity
- PMID: 33144315
- PMCID: PMC7646530
- DOI: 10.1128/mSystems.01045-20
Large-Scale Metagenome Assembly Reveals Novel Animal-Associated Microbial Genomes, Biosynthetic Gene Clusters, and Other Genetic Diversity
Abstract
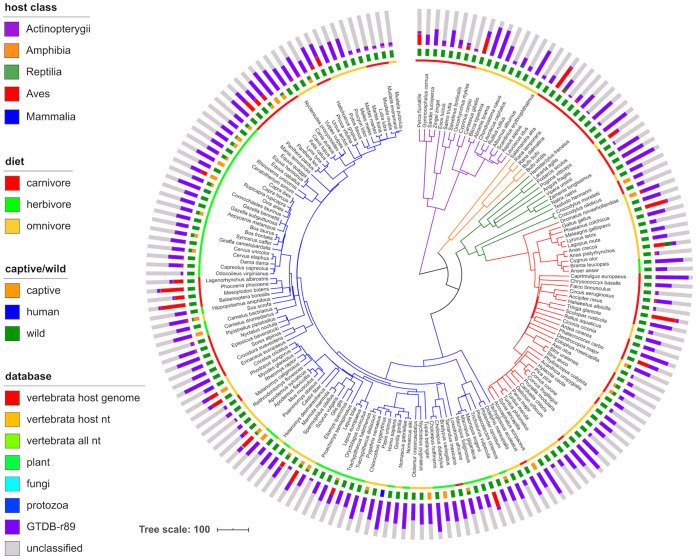
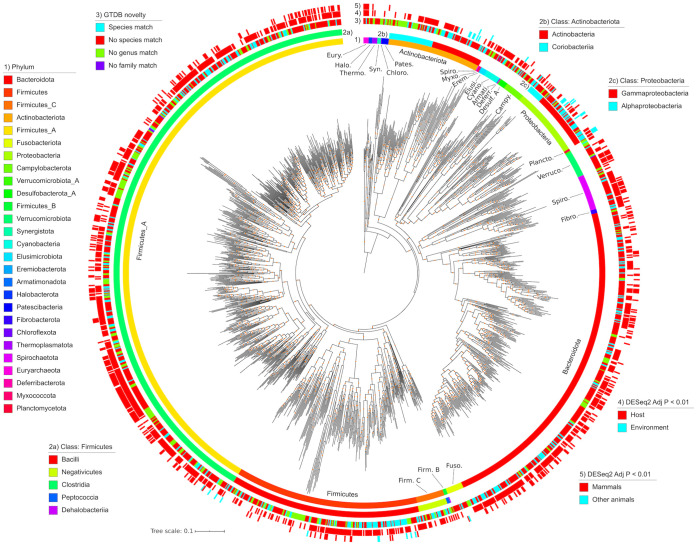
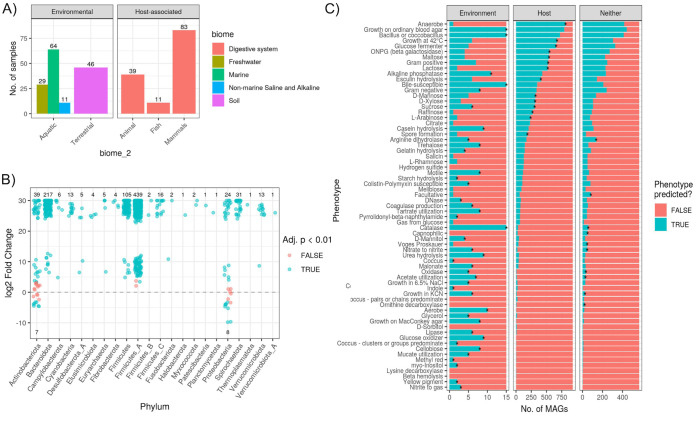
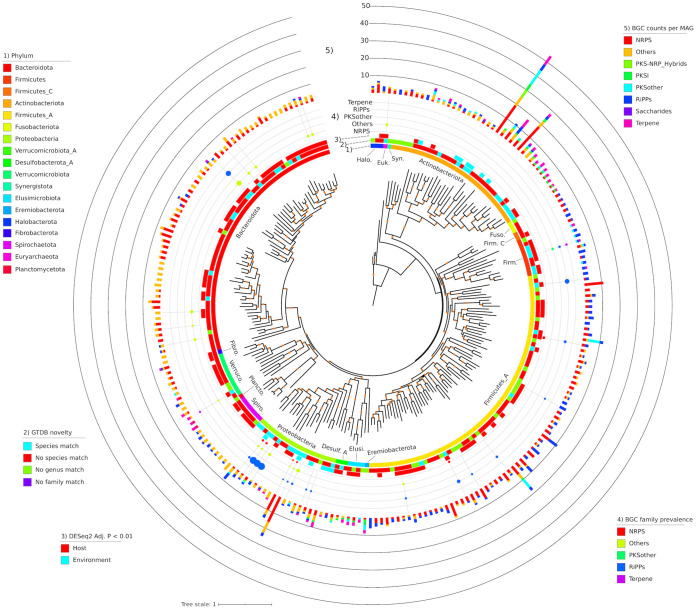
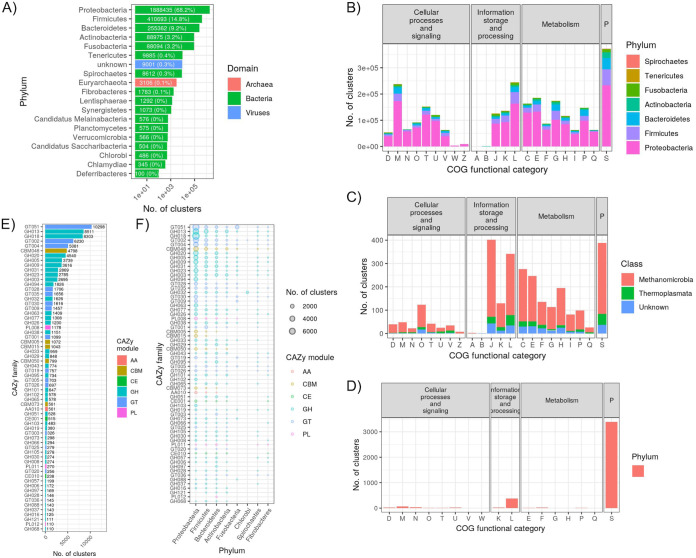
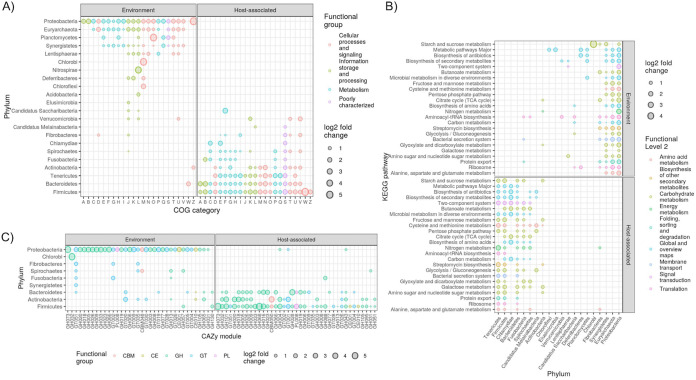
Large-scale metagenome assemblies of human microbiomes have produced a vast catalogue of previously unseen microbial genomes; however, comparatively few microbial genomes derive from other vertebrates. Here, we generated 5,596 metagenome-assembled genomes (MAGs) from the gut metagenomes of 180 predominantly wild animal species representing 5 classes, in addition to 14 existing animal gut metagenome data sets. The MAGs comprised 1,522 species-level genome bins (SGBs), most of which were novel at the species, genus, or family level, and the majority were enriched in host versus environment metagenomes. Many traits distinguished SGBs enriched in host or environmental biomes, including the number of antimicrobial resistance genes. We identified 1,986 diverse biosynthetic gene clusters; only 23 clustered with any MIBiG database references. Gene-based assembly revealed tremendous gene diversity, much of it host or environment specific. Our MAG and gene data sets greatly expand the microbial genome repertoire and provide a broad view of microbial adaptations to the vertebrate gut.IMPORTANCE Microbiome studies on a select few mammalian species (e.g., humans, mice, and cattle) have revealed a great deal of novel genomic diversity in the gut microbiome. However, little is known of the microbial diversity in the gut of other vertebrates. We studied the gut microbiomes of a large set of mostly wild animal species consisting of mammals, birds, reptiles, amphibians, and fish. Unfortunately, we found that existing reference databases commonly used for metagenomic analyses failed to capture the microbiome diversity among vertebrates. To increase database representation, we applied advanced metagenome assembly methods to our animal gut data and to many public gut metagenome data sets that had not been used to obtain microbial genomes. Our resulting genome and gene cluster collections comprised a great deal of novel taxonomic and genomic diversity, which we extensively characterized. Our findings substantially expand what is known of microbial genomic diversity in the vertebrate gut.
Keywords: animal microbiome; antimicrobial resistance; biosynthetic gene cluster; gut; metagenome assembly; novel diversity; vertebrate-microbe.
Copyright © 2020 Youngblut et al.
Figures
Similar articles
-
Taxonomic, Genomic, and Functional Variation in the Gut Microbiomes of Wild Spotted Hyenas Across 2 Decades of Study.mSystems. 2023 Feb 23;8(1):e0096522. doi: 10.1128/msystems.00965-22. Epub 2022 Dec 19. mSystems. 2023. PMID: 36533929 Free PMC article.
-
Mining metagenomic data to gain a new insight into the gut microbial biosynthetic potential in placental mammals.Microbiol Spectr. 2024 Oct 3;12(10):e0086424. doi: 10.1128/spectrum.00864-24. Epub 2024 Aug 20. Microbiol Spectr. 2024. PMID: 39162518 Free PMC article.
-
A catalog of microbial genes and metagenome-assembled genomes from the quail gut microbiome.Poult Sci. 2023 Oct;102(10):102931. doi: 10.1016/j.psj.2023.102931. Epub 2023 Jul 9. Poult Sci. 2023. PMID: 37499616 Free PMC article.
-
Global landscape of gut microbiome diversity and antibiotic resistomes across vertebrates.Sci Total Environ. 2022 Sep 10;838(Pt 2):156178. doi: 10.1016/j.scitotenv.2022.156178. Epub 2022 May 23. Sci Total Environ. 2022. PMID: 35618126 Review.
-
A review of computational tools for generating metagenome-assembled genomes from metagenomic sequencing data.Comput Struct Biotechnol J. 2021 Nov 23;19:6301-6314. doi: 10.1016/j.csbj.2021.11.028. eCollection 2021. Comput Struct Biotechnol J. 2021. PMID: 34900140 Free PMC article. Review.
Cited by
-
Complete mitochondrial genome and phylogenetic analysis of Chloris chloris (Passeriformes: Fringillidae).Mitochondrial DNA B Resour. 2024 Oct 1;9(10):1327-1330. doi: 10.1080/23802359.2024.2410468. eCollection 2024. Mitochondrial DNA B Resour. 2024. PMID: 39359379 Free PMC article.
-
Exploring microbial diversity and biosynthetic potential in zoo and wildlife animal microbiomes.Nat Commun. 2024 Sep 26;15(1):8263. doi: 10.1038/s41467-024-52669-9. Nat Commun. 2024. PMID: 39327429 Free PMC article.
-
Advancing Source Tracking: Systematic Review and Source-Specific Genome Database Curation of Fecally Shed Prokaryotes.Environ Sci Technol Lett. 2024 Aug 7;11(9):931-939. doi: 10.1021/acs.estlett.4c00233. eCollection 2024 Sep 10. Environ Sci Technol Lett. 2024. PMID: 39280079 Free PMC article.
-
Turning the needle into the haystack: Culture-independent amplification of complex microbial genomes directly from their native environment.PLoS Pathog. 2024 Sep 12;20(9):e1012418. doi: 10.1371/journal.ppat.1012418. eCollection 2024 Sep. PLoS Pathog. 2024. PMID: 39264872 Free PMC article. Review.
-
Advances in lasso peptide discovery, biosynthesis, and function.Trends Genet. 2024 Nov;40(11):950-968. doi: 10.1016/j.tig.2024.08.002. Epub 2024 Aug 31. Trends Genet. 2024. PMID: 39218755 Review.
References
-
- Zou Y, Xue W, Luo G, Deng Z, Qin P, Guo R, Sun H, Xia Y, Liang S, Dai Y, Wan D, Jiang R, Su L, Feng Q, Jie Z, Guo T, Xia Z, Liu C, Yu J, Lin Y, Tang S, Huo G, Xu X, Hou Y, Liu X, Wang J, Yang H, Kristiansen K, Li J, Jia H, Xiao L. 2019. 1,520 reference genomes from cultivated human gut bacteria enable functional microbiome analyses. Nat Biotechnol 37:179–185. doi:10.1038/s41587-018-0008-8. - DOI - PMC - PubMed
-
- Forster SC, Kumar N, Anonye BO, Almeida A, Viciani E, Stares MD, Dunn M, Mkandawire TT, Zhu A, Shao Y, Pike LJ, Louie T, Browne HP, Mitchell AL, Neville BA, Finn RD, Lawley TD. 2019. A human gut bacterial genome and culture collection for improved metagenomic analyses. Nat Biotechnol 37:186–192. doi:10.1038/s41587-018-0009-7. - DOI - PMC - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
