

mixOmics: An R package for 'omics feature selection and multiple data integration
- PMID: 29099853
- PMCID: PMC5687754
- DOI: 10.1371/journal.pcbi.1005752
mixOmics: An R package for 'omics feature selection and multiple data integration
Abstract
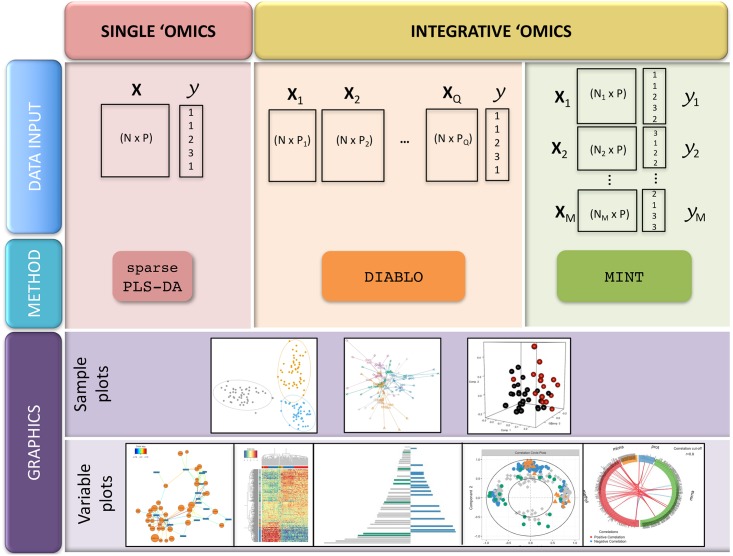
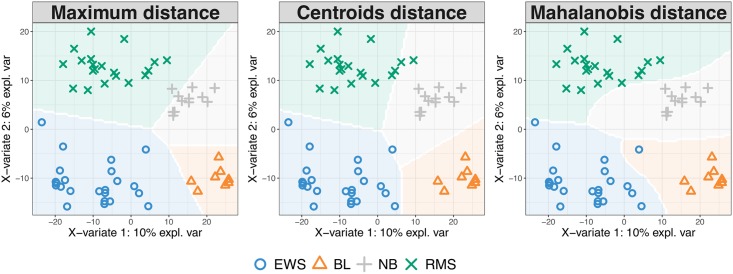
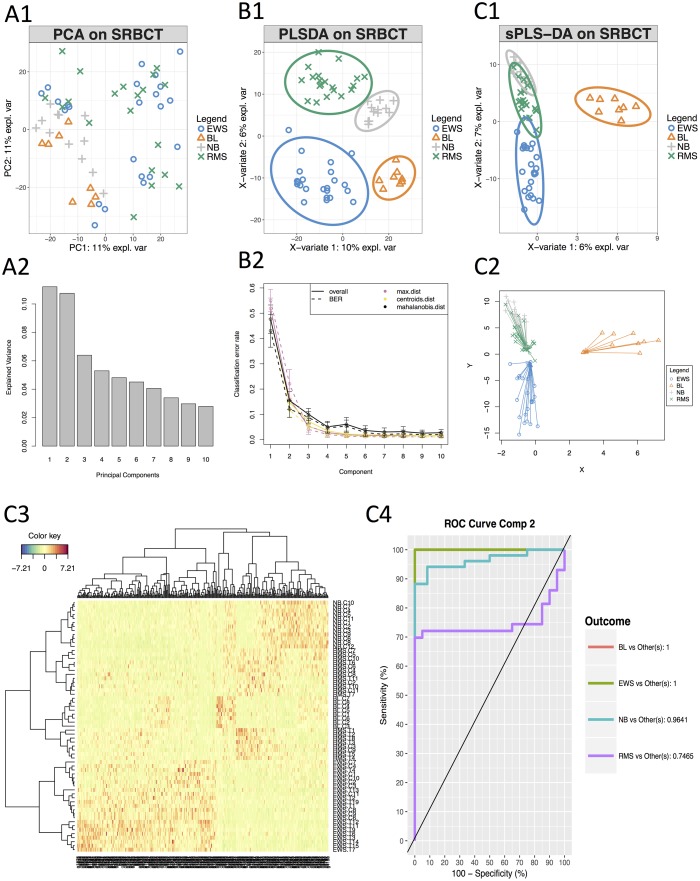
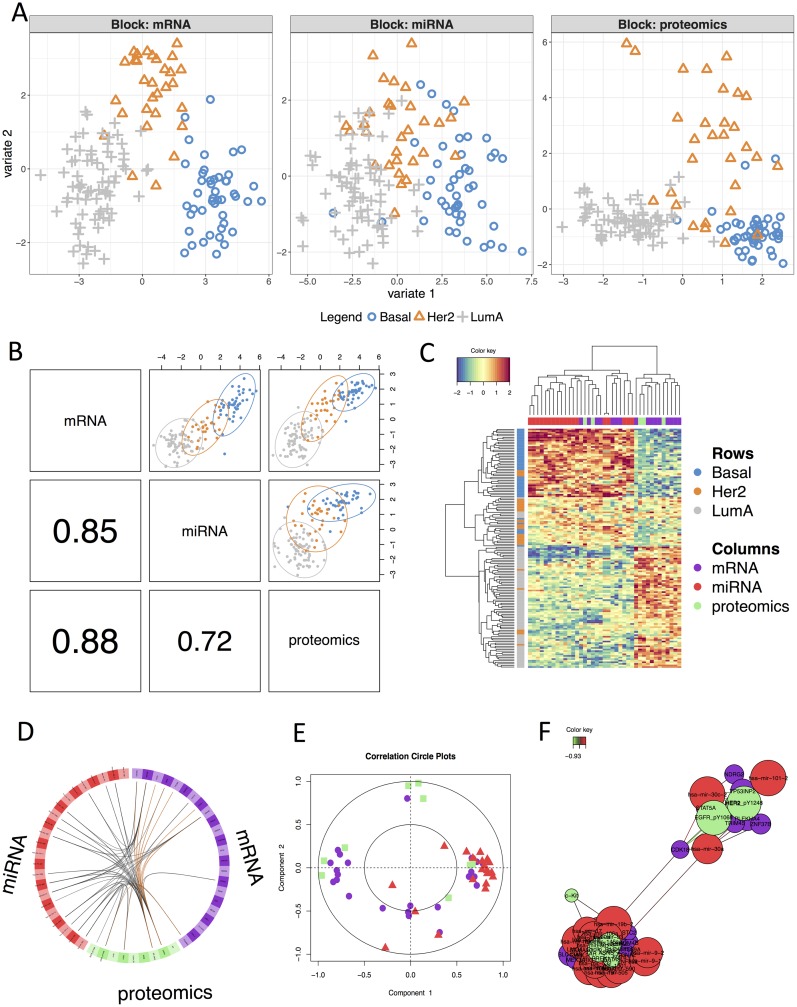
The advent of high throughput technologies has led to a wealth of publicly available 'omics data coming from different sources, such as transcriptomics, proteomics, metabolomics. Combining such large-scale biological data sets can lead to the discovery of important biological insights, provided that relevant information can be extracted in a holistic manner. Current statistical approaches have been focusing on identifying small subsets of molecules (a 'molecular signature') to explain or predict biological conditions, but mainly for a single type of 'omics. In addition, commonly used methods are univariate and consider each biological feature independently. We introduce mixOmics, an R package dedicated to the multivariate analysis of biological data sets with a specific focus on data exploration, dimension reduction and visualisation. By adopting a systems biology approach, the toolkit provides a wide range of methods that statistically integrate several data sets at once to probe relationships between heterogeneous 'omics data sets. Our recent methods extend Projection to Latent Structure (PLS) models for discriminant analysis, for data integration across multiple 'omics data or across independent studies, and for the identification of molecular signatures. We illustrate our latest mixOmics integrative frameworks for the multivariate analyses of 'omics data available from the package.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
Similar articles
-
Multivariate Analysis with the R Package mixOmics.Methods Mol Biol. 2023;2426:333-359. doi: 10.1007/978-1-0716-1967-4_15. Methods Mol Biol. 2023. PMID: 36308696
-
A powerful framework for an integrative study with heterogeneous omics data: from univariate statistics to multi-block analysis.Brief Bioinform. 2021 May 20;22(3):bbaa166. doi: 10.1093/bib/bbaa166. Brief Bioinform. 2021. PMID: 32778869
-
Translational Metabolomics of Head Injury: Exploring Dysfunctional Cerebral Metabolism with Ex Vivo NMR Spectroscopy-Based Metabolite Quantification.In: Kobeissy FH, editor. Brain Neurotrauma: Molecular, Neuropsychological, and Rehabilitation Aspects. Boca Raton (FL): CRC Press/Taylor & Francis; 2015. Chapter 25. In: Kobeissy FH, editor. Brain Neurotrauma: Molecular, Neuropsychological, and Rehabilitation Aspects. Boca Raton (FL): CRC Press/Taylor & Francis; 2015. Chapter 25. PMID: 26269925 Free Books & Documents. Review.
-
Multi-omics integration in biomedical research - A metabolomics-centric review.Anal Chim Acta. 2021 Jan 2;1141:144-162. doi: 10.1016/j.aca.2020.10.038. Epub 2020 Oct 22. Anal Chim Acta. 2021. PMID: 33248648 Free PMC article. Review.
-
DIABLO: an integrative approach for identifying key molecular drivers from multi-omics assays.Bioinformatics. 2019 Sep 1;35(17):3055-3062. doi: 10.1093/bioinformatics/bty1054. Bioinformatics. 2019. PMID: 30657866 Free PMC article.
Cited by
-
The BNT162b2 mRNA vaccine demonstrates reduced age-associated TH1 support in vitro and in vivo.iScience. 2024 Sep 26;27(11):111055. doi: 10.1016/j.isci.2024.111055. eCollection 2024 Nov 15. iScience. 2024. PMID: 39569372 Free PMC article.
-
Targeting AXL cellular networks in kidney fibrosis.Front Immunol. 2024 Nov 4;15:1446672. doi: 10.3389/fimmu.2024.1446672. eCollection 2024. Front Immunol. 2024. PMID: 39559366 Free PMC article.
-
Comparative transcriptomic analyses of diploid and tetraploid citrus reveal how ploidy level influences salt stress tolerance.Front Plant Sci. 2024 Oct 30;15:1469115. doi: 10.3389/fpls.2024.1469115. eCollection 2024. Front Plant Sci. 2024. PMID: 39544537 Free PMC article.
-
Analysis of blood metabolite characteristics at birth in preterm infants with bronchopulmonary dysplasia: an observational cohort study.Front Pediatr. 2024 Oct 31;12:1474381. doi: 10.3389/fped.2024.1474381. eCollection 2024. Front Pediatr. 2024. PMID: 39544337 Free PMC article.
-
Diagnosis and prognosis prediction of gastric cancer by high-performance serum lipidome fingerprints.EMBO Mol Med. 2024 Dec;16(12):3089-3112. doi: 10.1038/s44321-024-00169-0. Epub 2024 Nov 14. EMBO Mol Med. 2024. PMID: 39543322 Free PMC article.
References
-
- Lê Cao KA, Rohart F, Gonzalez I, Déjean S, Gautier B, Bartolo F, et al. mixOmics: Omics Data Integration Project; 2017. Available from: https://CRAN.R-project.org/package=mixOmics.
-
- Boulesteix AL, Strimmer K. Partial least squares: a versatile tool for the analysis of high-dimensional genomic data. Brief Bioinform. 2007;8(1):32–44. doi: 10.1093/bib/bbl016 - DOI - PubMed
-
- Meng C, Zeleznik OA, Thallinger GG, Kuster B, Gholami AM, Culhane AC. Dimension reduction techniques for the integrative analysis of multi-omics data. Briefings in bioinformatics. 2016; p. bbv108. doi: 10.1093/bib/bbv108 - DOI - PMC - PubMed
-
- Labus JS, Van Horn JD, Gupta A, Alaverdyan M, Torgerson C, Ashe-McNalley C, et al. Multivariate morphological brain signatures predict patients with chronic abdominal pain from healthy control subjects. Pain. 2015;156(8):1545–1554. doi: 10.1097/j.pain.0000000000000196 - DOI - PMC - PubMed
-
- Cook JA, Chandramouli GV, Anver MR, Sowers AL, Thetford A, Krausz KW, et al. Mass Spectrometry–Based Metabolomics Identifies Longitudinal Urinary Metabolite Profiles Predictive of Radiation-Induced Cancer. Cancer research. 2016;76(6):1569–1577. doi: 10.1158/0008-5472.CAN-15-2416 - DOI - PMC - PubMed
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
