

MrTADFinder: A network modularity based approach to identify topologically associating domains in multiple resolutions
- PMID: 28742097
- PMCID: PMC5546724
- DOI: 10.1371/journal.pcbi.1005647
MrTADFinder: A network modularity based approach to identify topologically associating domains in multiple resolutions
Abstract
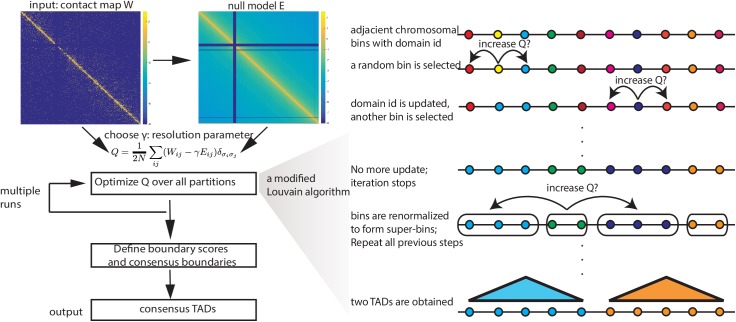
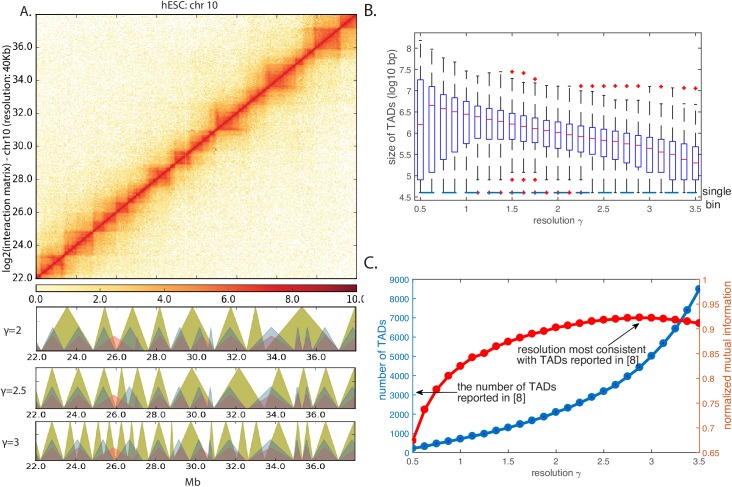
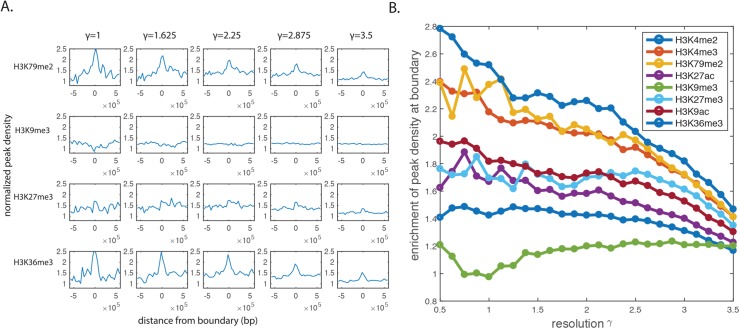
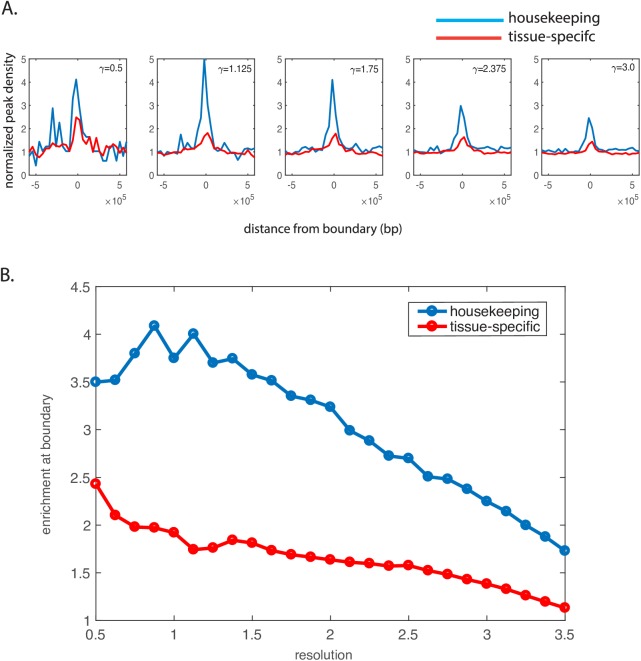
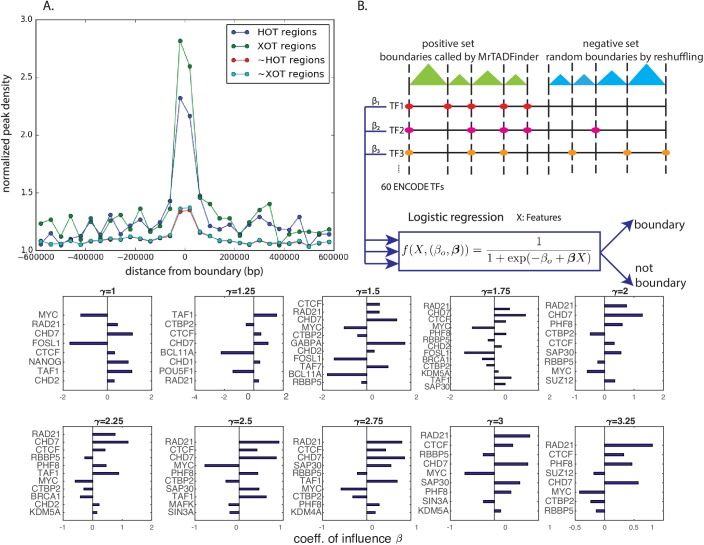
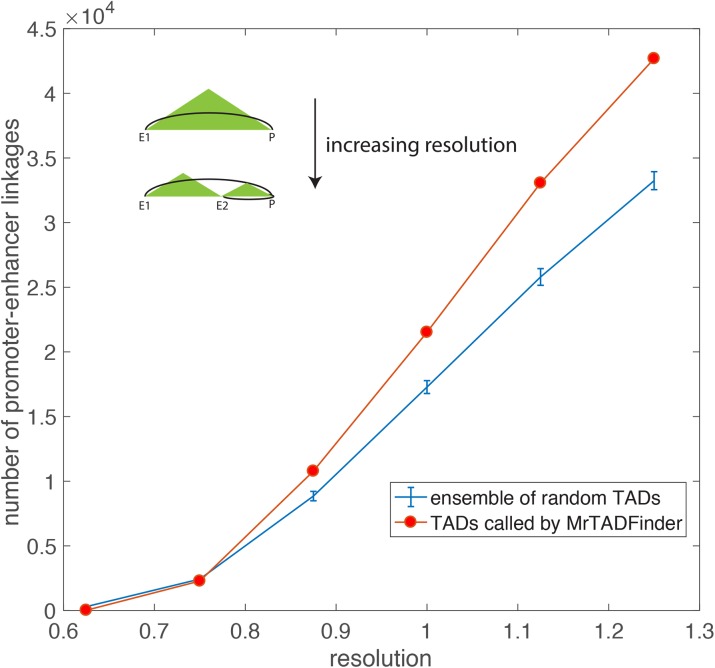
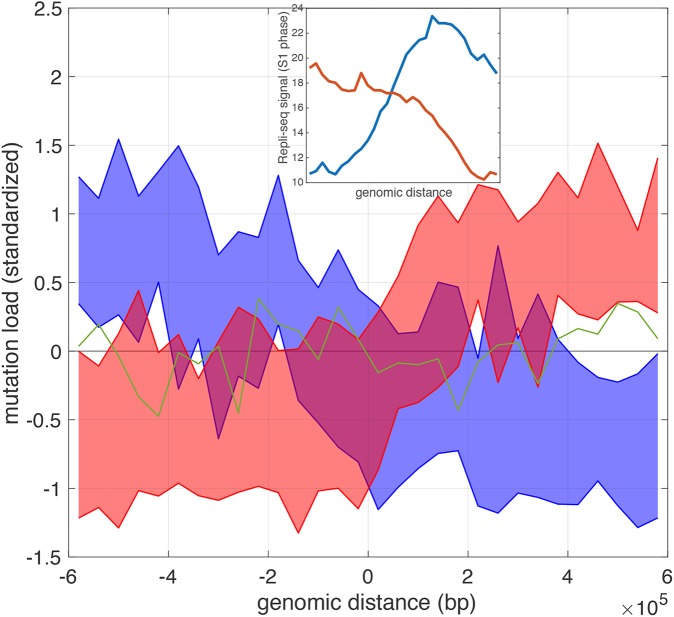
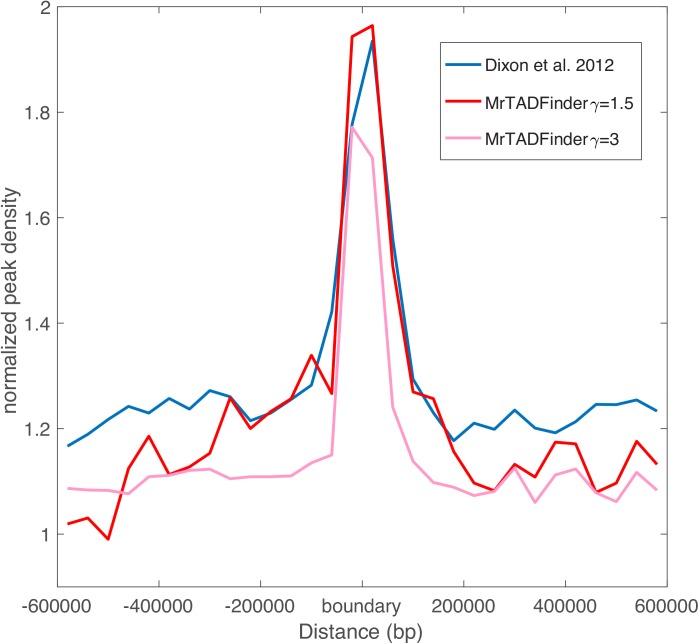
Genome-wide proximity ligation based assays such as Hi-C have revealed that eukaryotic genomes are organized into structural units called topologically associating domains (TADs). From a visual examination of the chromosomal contact map, however, it is clear that the organization of the domains is not simple or obvious. Instead, TADs exhibit various length scales and, in many cases, a nested arrangement. Here, by exploiting the resemblance between TADs in a chromosomal contact map and densely connected modules in a network, we formulate TAD identification as a network optimization problem and propose an algorithm, MrTADFinder, to identify TADs from intra-chromosomal contact maps. MrTADFinder is based on the network-science concept of modularity. A key component of it is deriving an appropriate background model for contacts in a random chain, by numerically solving a set of matrix equations. The background model preserves the observed coverage of each genomic bin as well as the distance dependence of the contact frequency for any pair of bins exhibited by the empirical map. Also, by introducing a tunable resolution parameter, MrTADFinder provides a self-consistent approach for identifying TADs at different length scales, hence the acronym "Mr" standing for Multiple Resolutions. We then apply MrTADFinder to various Hi-C datasets. The identified domain boundaries are marked by characteristic signatures in chromatin marks and transcription factors (TF) that are consistent with earlier work. Moreover, by calling TADs at different length scales, we observe that boundary signatures change with resolution, with different chromatin features having different characteristic length scales. Furthermore, we report an enrichment of HOT (high-occupancy target) regions near TAD boundaries and investigate the role of different TFs in determining boundaries at various resolutions. To further explore the interplay between TADs and epigenetic marks, as tumor mutational burden is known to be coupled to chromatin structure, we examine how somatic mutations are distributed across boundaries and find a clear stepwise pattern. Overall, MrTADFinder provides a novel computational framework to explore the multi-scale structures in Hi-C contact maps.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
Similar articles
-
Methods for the Analysis of Topologically Associating Domains (TADs).Methods Mol Biol. 2022;2301:39-59. doi: 10.1007/978-1-0716-1390-0_3. Methods Mol Biol. 2022. PMID: 34415530
-
Identifying topologically associating domains using differential kernels.PLoS Comput Biol. 2024 Jul 15;20(7):e1012221. doi: 10.1371/journal.pcbi.1012221. eCollection 2024 Jul. PLoS Comput Biol. 2024. PMID: 39008525 Free PMC article.
-
A Comparison of Topologically Associating Domain Callers Based on Hi-C Data.IEEE/ACM Trans Comput Biol Bioinform. 2023 Jan-Feb;20(1):15-29. doi: 10.1109/TCBB.2022.3147805. Epub 2023 Feb 3. IEEE/ACM Trans Comput Biol Bioinform. 2023. PMID: 35104223
-
TADs and Their Borders: Free Movement or Building a Wall?J Mol Biol. 2020 Feb 7;432(3):643-652. doi: 10.1016/j.jmb.2019.11.025. Epub 2019 Dec 27. J Mol Biol. 2020. PMID: 31887284 Review.
-
Are TADs supercoiled?Nucleic Acids Res. 2019 Jan 25;47(2):521-532. doi: 10.1093/nar/gky1091. Nucleic Acids Res. 2019. PMID: 30395328 Free PMC article. Review.
Cited by
-
A subset of topologically associating domains fold into mesoscale core-periphery networks.Sci Rep. 2019 Jul 2;9(1):9526. doi: 10.1038/s41598-019-45457-9. Sci Rep. 2019. PMID: 31266973 Free PMC article.
-
Network Modeling in Biology: Statistical Methods for Gene and Brain Networks.Stat Sci. 2021 Feb;36(1):89-108. doi: 10.1214/20-sts792. Stat Sci. 2021. PMID: 34305304 Free PMC article.
-
CASPIAN: A method to identify chromatin topological associated domains based on spatial density cluster.Comput Struct Biotechnol J. 2022 Sep 5;20:4816-4824. doi: 10.1016/j.csbj.2022.08.059. eCollection 2022. Comput Struct Biotechnol J. 2022. PMID: 36147659 Free PMC article.
-
NETWORK MODELLING OF TOPOLOGICAL DOMAINS USING HI-C DATA.Ann Appl Stat. 2019 Sep;13(3):1511-1536. doi: 10.1214/19-aoas1244. Epub 2019 Oct 17. Ann Appl Stat. 2019. PMID: 32968472 Free PMC article.
-
Mapping robust multiscale communities in chromosome contact networks.Sci Rep. 2023 Aug 10;13(1):12979. doi: 10.1038/s41598-023-39522-7. Sci Rep. 2023. PMID: 37563218 Free PMC article.
References
-
- Dekker J, Marti-Renom MA, Mirny LA. Exploring the three-dimensional organization of genomes: interpreting chromatin interaction data. Nat Rev Genet. 2013;14: 390–403. doi: 10.1038/nrg3454 - DOI - PMC - PubMed
-
- Risca VI, Greenleaf WJ. Unraveling the 3D genome: genomics tools for multiscale exploration. Trends Genet. 2015;31: 357–372. doi: 10.1016/j.tig.2015.03.010 - DOI - PMC - PubMed
-
- Rowley MJ, Corces VG. The three-dimensional genome: principles and roles of long-distance interactions. Curr Opin Cell Biol. 2016;40: 8–14. doi: 10.1016/j.ceb.2016.01.009 - DOI - PMC - PubMed
-
- Bonev B, Cavalli G. Organization and function of the 3D genome. Nat Rev Genet. 2016;17: 661–678. doi: 10.1038/nrg.2016.112 - DOI - PubMed
-
- Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, et al. Comprehensive Mapping of Long-Range Interactions Reveals Folding Principles of the Human Genome. Science. 2009;326: 289–293. doi: 10.1126/science.1181369 - DOI - PMC - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
