

3D structures of individual mammalian genomes studied by single-cell Hi-C
- PMID: 28289288
- PMCID: PMC5385134
- DOI: 10.1038/nature21429
3D structures of individual mammalian genomes studied by single-cell Hi-C
Abstract
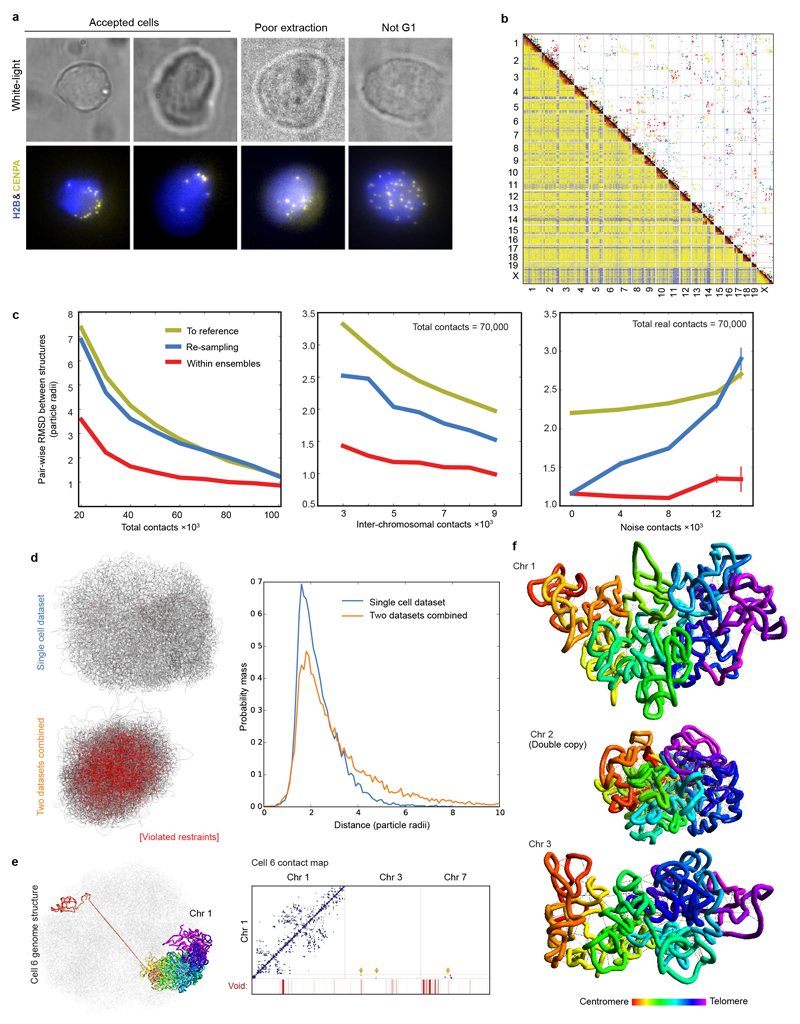
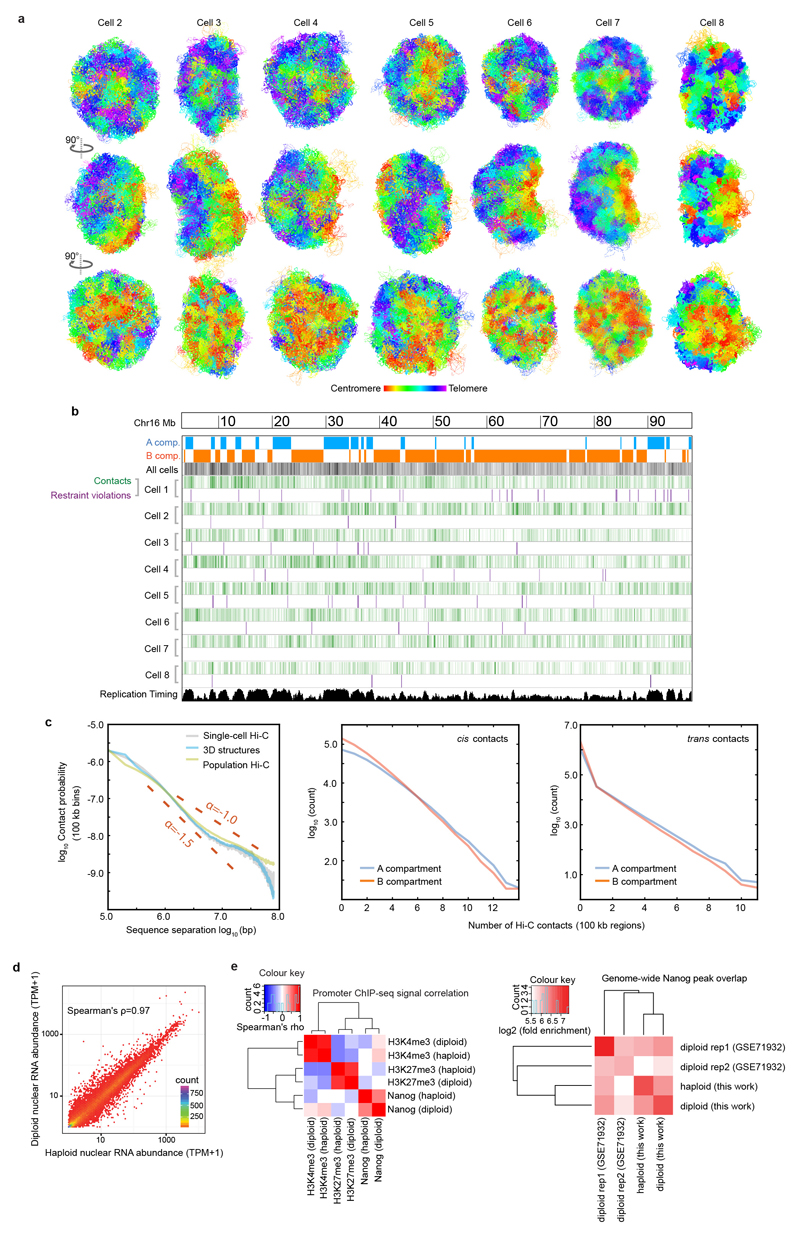
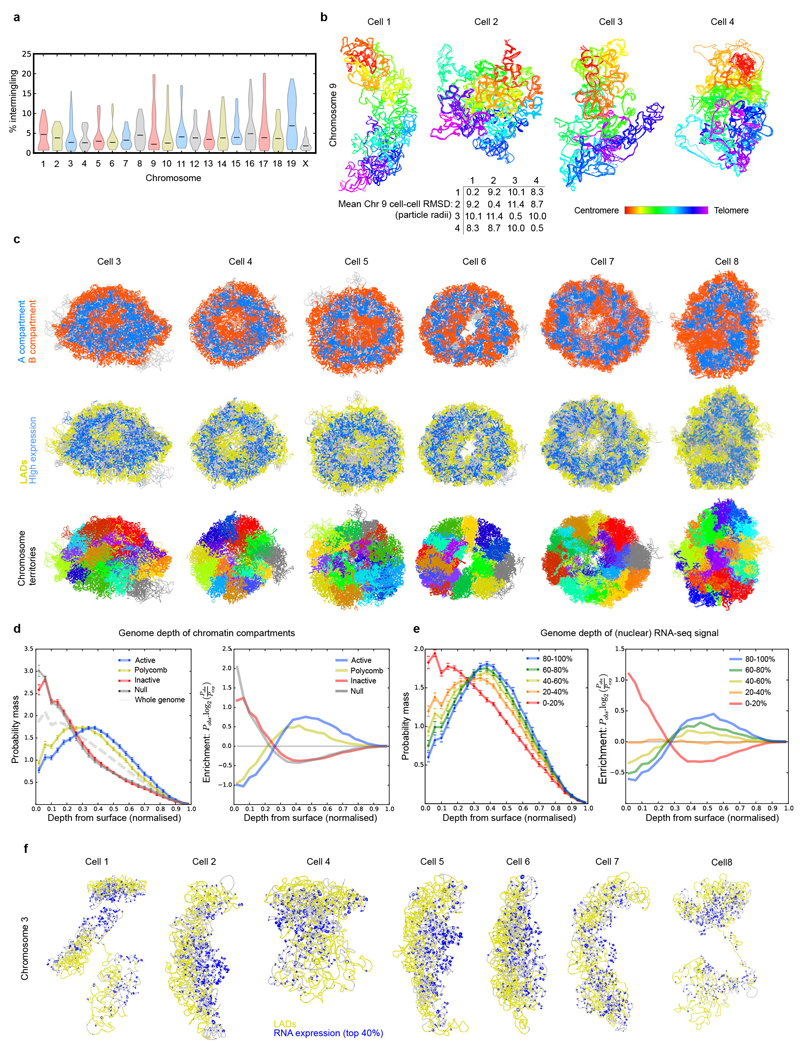
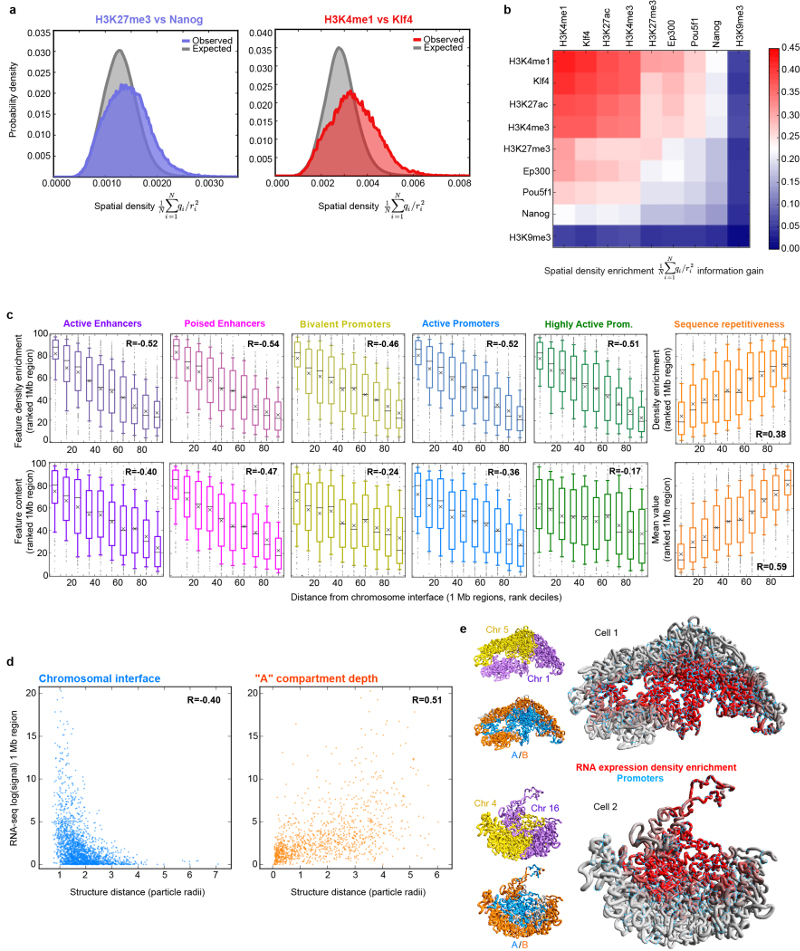
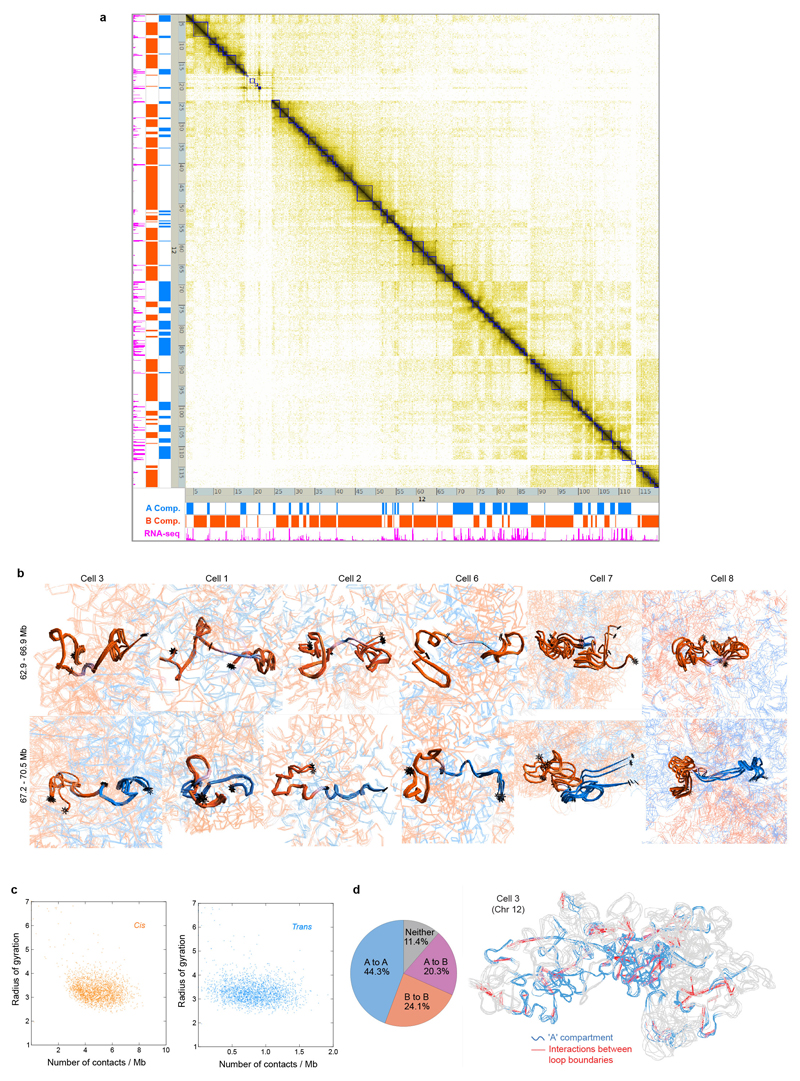
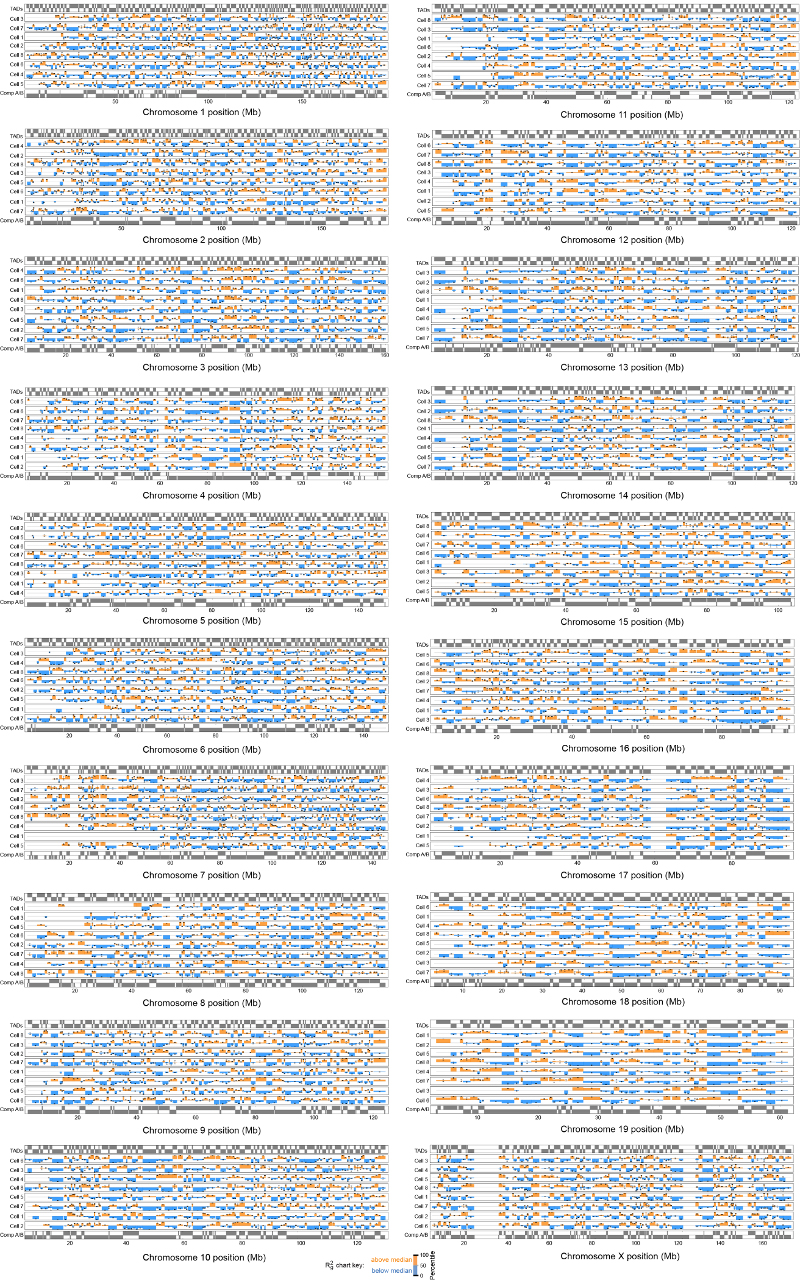
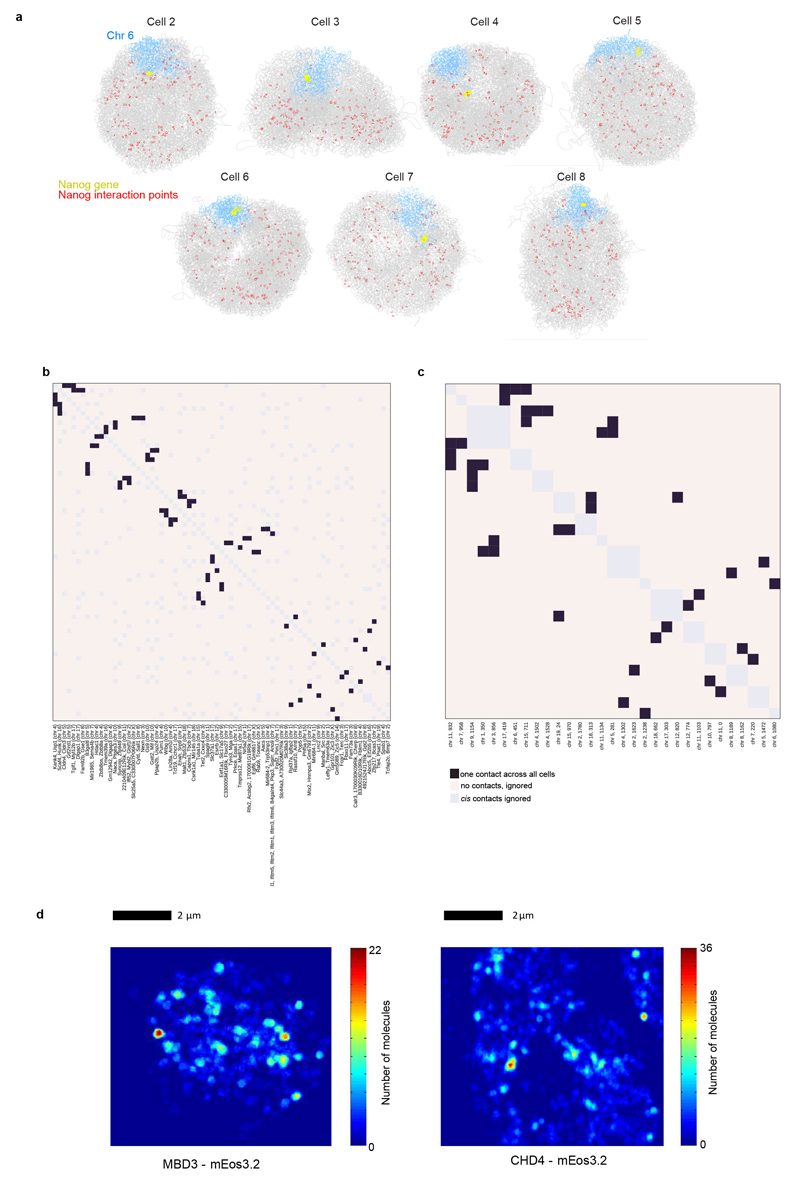
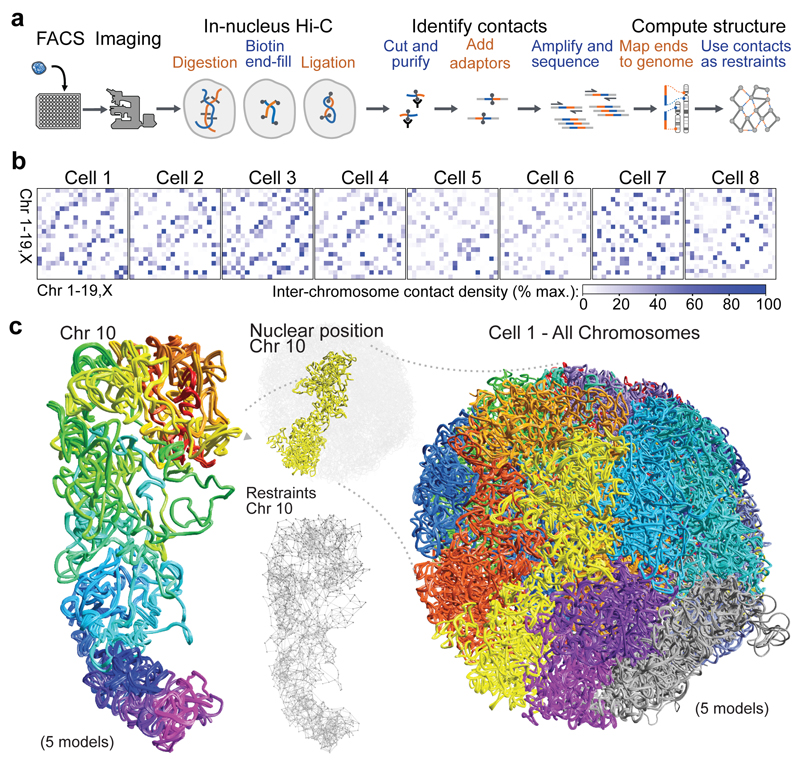
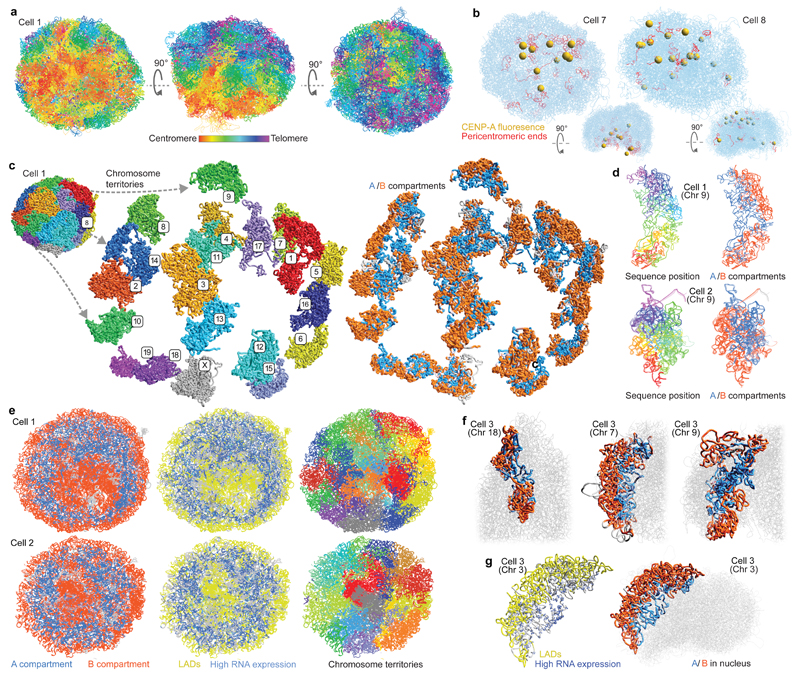
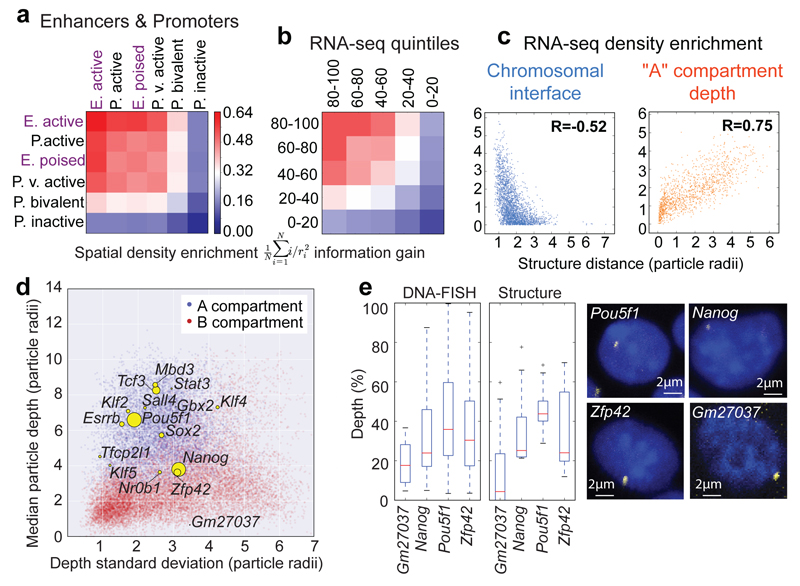
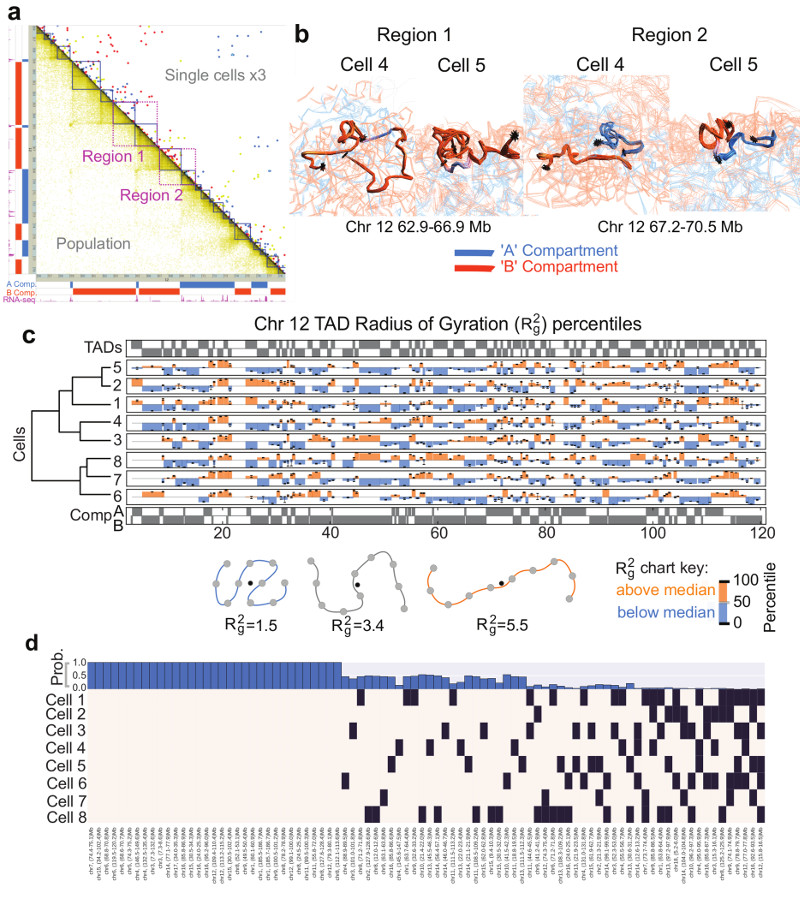
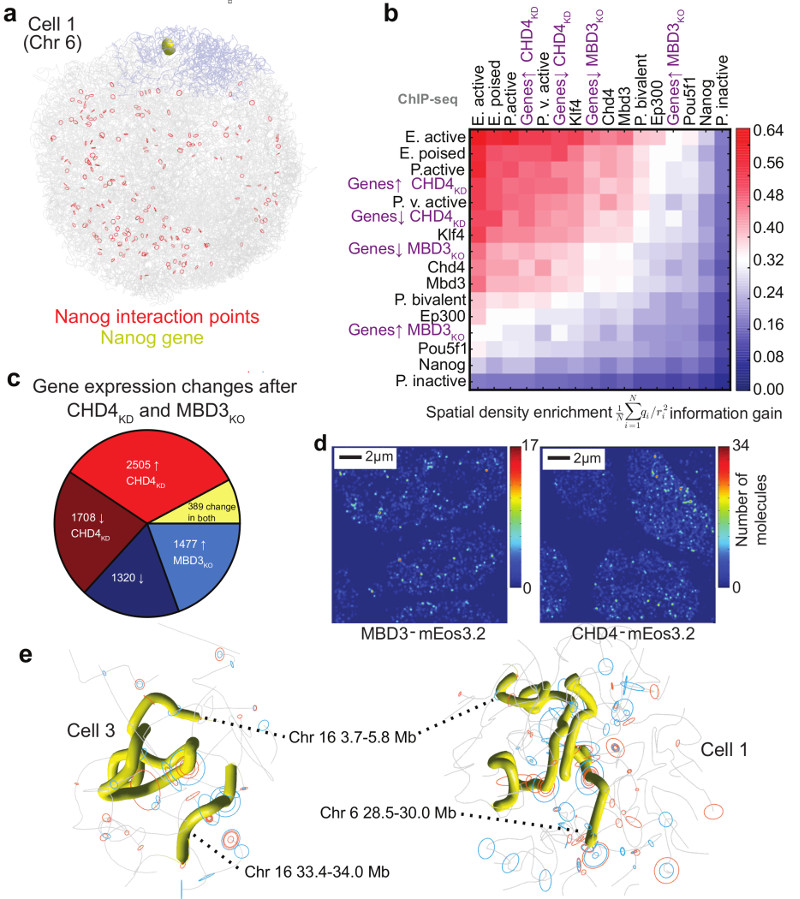
The folding of genomic DNA from the beads-on-a-string-like structure of nucleosomes into higher-order assemblies is crucially linked to nuclear processes. Here we calculate 3D structures of entire mammalian genomes using data from a new chromosome conformation capture procedure that allows us to first image and then process single cells. The technique enables genome folding to be examined at a scale of less than 100 kb, and chromosome structures to be validated. The structures of individual topological-associated domains and loops vary substantially from cell to cell. By contrast, A and B compartments, lamina-associated domains and active enhancers and promoters are organized in a consistent way on a genome-wide basis in every cell, suggesting that they could drive chromosome and genome folding. By studying genes regulated by pluripotency factor and nucleosome remodelling deacetylase (NuRD), we illustrate how the determination of single-cell genome structure provides a new approach for investigating biological processes.
Conflict of interest statement
The authors declare no competing interests.
Figures

Comment in
-
Genome Organization: Zooming in on nuclear organization.Nat Rev Mol Cell Biol. 2017 May;18(5):275. doi: 10.1038/nrm.2017.28. Epub 2017 Mar 22. Nat Rev Mol Cell Biol. 2017. PMID: 28327555 No abstract available.
-
Genome organization: Zooming in on nuclear organization.Nat Rev Genet. 2017 May;18(5):269. doi: 10.1038/nrg.2017.23. Epub 2017 Mar 27. Nat Rev Genet. 2017. PMID: 28344343 No abstract available.
Similar articles
-
Cell-cycle dynamics of chromosomal organization at single-cell resolution.Nature. 2017 Jul 5;547(7661):61-67. doi: 10.1038/nature23001. Nature. 2017. PMID: 28682332 Free PMC article.
-
Ctbp2 Modulates NuRD-Mediated Deacetylation of H3K27 and Facilitates PRC2-Mediated H3K27me3 in Active Embryonic Stem Cell Genes During Exit from Pluripotency.Stem Cells. 2015 Aug;33(8):2442-55. doi: 10.1002/stem.2046. Epub 2015 May 26. Stem Cells. 2015. PMID: 25944056
-
NuRD-interacting protein ZFP296 regulates genome-wide NuRD localization and differentiation of mouse embryonic stem cells.Nat Commun. 2018 Nov 2;9(1):4588. doi: 10.1038/s41467-018-07063-7. Nat Commun. 2018. PMID: 30389936 Free PMC article.
-
Unraveling the multiplex folding of nucleosome chains in higher order chromatin.Essays Biochem. 2019 Apr 23;63(1):109-121. doi: 10.1042/EBC20180066. Print 2019 Apr 23. Essays Biochem. 2019. PMID: 31015386 Review.
-
Nucleosome distribution and linker DNA: connecting nuclear function to dynamic chromatin structure.Biochem Cell Biol. 2011 Feb;89(1):24-34. doi: 10.1139/O10-139. Biochem Cell Biol. 2011. PMID: 21326360 Free PMC article. Review.
Cited by
-
Normalization and de-noising of single-cell Hi-C data with BandNorm and scVI-3D.Genome Biol. 2022 Oct 17;23(1):222. doi: 10.1186/s13059-022-02774-z. Genome Biol. 2022. PMID: 36253828 Free PMC article.
-
Epigenetic Modifications in Stress Response Genes Associated With Childhood Trauma.Front Psychiatry. 2019 Nov 8;10:808. doi: 10.3389/fpsyt.2019.00808. eCollection 2019. Front Psychiatry. 2019. PMID: 31780969 Free PMC article. Review.
-
Olfactory receptor genes make the case for inter-chromosomal interactions.Curr Opin Genet Dev. 2019 Apr;55:106-113. doi: 10.1016/j.gde.2019.07.004. Epub 2019 Sep 3. Curr Opin Genet Dev. 2019. PMID: 31491591 Free PMC article. Review.
-
Does multi-way, long-range chromatin contact data advance 3D genome reconstruction?BMC Bioinformatics. 2023 Feb 24;24(1):64. doi: 10.1186/s12859-023-05170-x. BMC Bioinformatics. 2023. PMID: 36829114 Free PMC article.
-
Analysis of the structural variability of topologically associated domains as revealed by Hi-C.NAR Genom Bioinform. 2020 Mar;2(1):lqz008. doi: 10.1093/nargab/lqz008. Epub 2019 Sep 30. NAR Genom Bioinform. 2020. PMID: 31687663 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
