

Chromatin extrusion explains key features of loop and domain formation in wild-type and engineered genomes
- PMID: 26499245
- PMCID: PMC4664323
- DOI: 10.1073/pnas.1518552112
Chromatin extrusion explains key features of loop and domain formation in wild-type and engineered genomes
Abstract
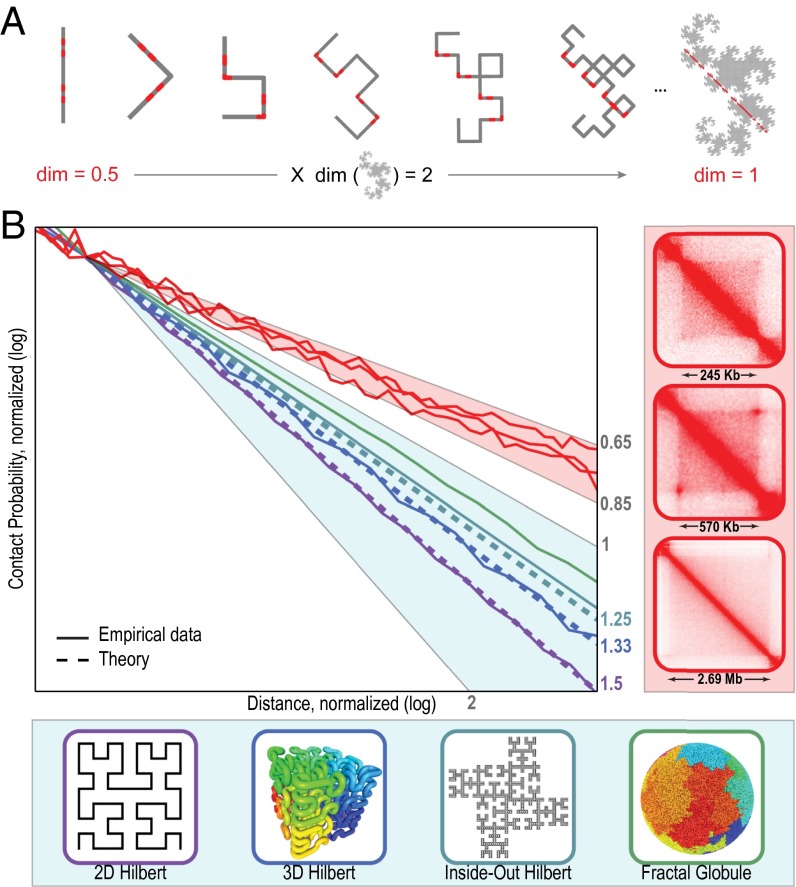
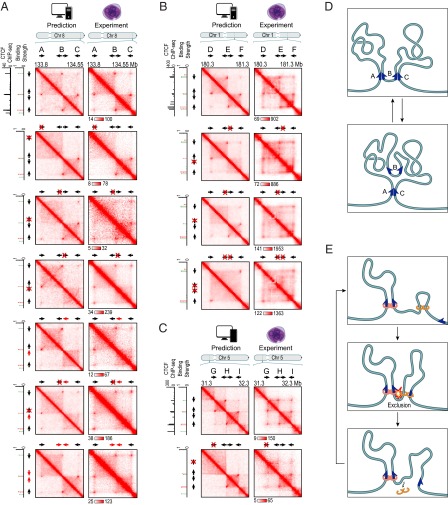
We recently used in situ Hi-C to create kilobase-resolution 3D maps of mammalian genomes. Here, we combine these maps with new Hi-C, microscopy, and genome-editing experiments to study the physical structure of chromatin fibers, domains, and loops. We find that the observed contact domains are inconsistent with the equilibrium state for an ordinary condensed polymer. Combining Hi-C data and novel mathematical theorems, we show that contact domains are also not consistent with a fractal globule. Instead, we use physical simulations to study two models of genome folding. In one, intermonomer attraction during polymer condensation leads to formation of an anisotropic "tension globule." In the other, CCCTC-binding factor (CTCF) and cohesin act together to extrude unknotted loops during interphase. Both models are consistent with the observed contact domains and with the observation that contact domains tend to form inside loops. However, the extrusion model explains a far wider array of observations, such as why loops tend not to overlap and why the CTCF-binding motifs at pairs of loop anchors lie in the convergent orientation. Finally, we perform 13 genome-editing experiments examining the effect of altering CTCF-binding sites on chromatin folding. The convergent rule correctly predicts the affected loops in every case. Moreover, the extrusion model accurately predicts in silico the 3D maps resulting from each experiment using only the location of CTCF-binding sites in the WT. Thus, we show that it is possible to disrupt, restore, and move loops and domains using targeted mutations as small as a single base pair.
Keywords: CRISPR; CTCF; chromatin loops; genome architecture; molecular dynamics.
Conflict of interest statement
The authors declare no conflict of interest.
Figures






Comment in
-
Predictive model of 3D domain formation via CTCF-mediated extrusion.Proc Natl Acad Sci U S A. 2015 Nov 24;112(47):14404-5. doi: 10.1073/pnas.1519849112. Epub 2015 Nov 13. Proc Natl Acad Sci U S A. 2015. PMID: 26567152 Free PMC article. No abstract available.
Similar articles
-
CTCF as a boundary factor for cohesin-mediated loop extrusion: evidence for a multi-step mechanism.Nucleus. 2020 Dec;11(1):132-148. doi: 10.1080/19491034.2020.1782024. Nucleus. 2020. PMID: 32631111 Free PMC article. Review.
-
Cohesin is positioned in mammalian genomes by transcription, CTCF and Wapl.Nature. 2017 Apr 27;544(7651):503-507. doi: 10.1038/nature22063. Epub 2017 Apr 19. Nature. 2017. PMID: 28424523 Free PMC article.
-
On the choreography of genome folding: A grand pas de deux of cohesin and CTCF.Curr Opin Cell Biol. 2021 Jun;70:84-90. doi: 10.1016/j.ceb.2020.12.001. Epub 2021 Feb 2. Curr Opin Cell Biol. 2021. PMID: 33545664 Review.
-
Depletion of the chromatin looping proteins CTCF and cohesin causes chromatin compaction: insight into chromatin folding by polymer modelling.PLoS Comput Biol. 2014 Oct 9;10(10):e1003877. doi: 10.1371/journal.pcbi.1003877. eCollection 2014 Oct. PLoS Comput Biol. 2014. PMID: 25299688 Free PMC article.
-
Chromatin jets define the properties of cohesin-driven in vivo loop extrusion.Mol Cell. 2022 Oct 20;82(20):3769-3780.e5. doi: 10.1016/j.molcel.2022.09.003. Epub 2022 Sep 30. Mol Cell. 2022. PMID: 36182691
Cited by
-
The 3D Genome in Brain Development: An Exploration of Molecular Mechanisms and Experimental Methods.Neurosci Insights. 2024 Oct 29;19:26331055241293455. doi: 10.1177/26331055241293455. eCollection 2024. Neurosci Insights. 2024. PMID: 39494115 Free PMC article. Review.
-
Alterations in Chromatin Folding Patterns in Cancer Variant-Enriched Loci.IEEE EMBS Int Conf Biomed Health Inform. 2019 May;2019:10.1109/bhi.2019.8834565. doi: 10.1109/bhi.2019.8834565. Epub 2019 Sep 12. IEEE EMBS Int Conf Biomed Health Inform. 2019. PMID: 34085045 Free PMC article.
-
The Molecular Impacts of Retrotransposons in Development and Diseases.Int J Mol Sci. 2023 Nov 16;24(22):16418. doi: 10.3390/ijms242216418. Int J Mol Sci. 2023. PMID: 38003607 Free PMC article. Review.
-
Cell-type-specific loops linked to RNA polymerase II elongation in human neural differentiation.Cell Genom. 2024 Aug 14;4(8):100606. doi: 10.1016/j.xgen.2024.100606. Epub 2024 Jul 10. Cell Genom. 2024. PMID: 38991604 Free PMC article.
-
Unraveling the mechanisms of chromatin fibril packaging.Nucleus. 2016 May 3;7(3):319-24. doi: 10.1080/19491034.2016.1190896. Nucleus. 2016. PMID: 27249516 Free PMC article.
References
-
- Kornberg RD, Lorch Y. Twenty-five years of the nucleosome, fundamental particle of the eukaryote chromosome. Cell. 1999;98(3):285–294. - PubMed
-
- Hansen JC. Conformational dynamics of the chromatin fiber in solution: Determinants, mechanisms, and functions. Annu Rev Biophys Biomol Struct. 2002;31(1):361–392. - PubMed
-
- Hartwig M. The size of independently supercoiled domains in nuclear DNA from normal human lymphocytes and leukemic lymphoblasts. Biochim Biophys Acta. 1982;698(2):214–217. - PubMed
-
- Zehnbauer BA, Vogelstein B. Supercoiled loops and the organization of replication and transcription in eukaryotes. BioEssays. 1985;2(2):52–54.
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
