

Arrhythmogenesis in a catecholaminergic polymorphic ventricular tachycardia mutation that depresses ryanodine receptor function
- PMID: 25775566
- PMCID: PMC4386375
- DOI: 10.1073/pnas.1419795112
Arrhythmogenesis in a catecholaminergic polymorphic ventricular tachycardia mutation that depresses ryanodine receptor function
Abstract
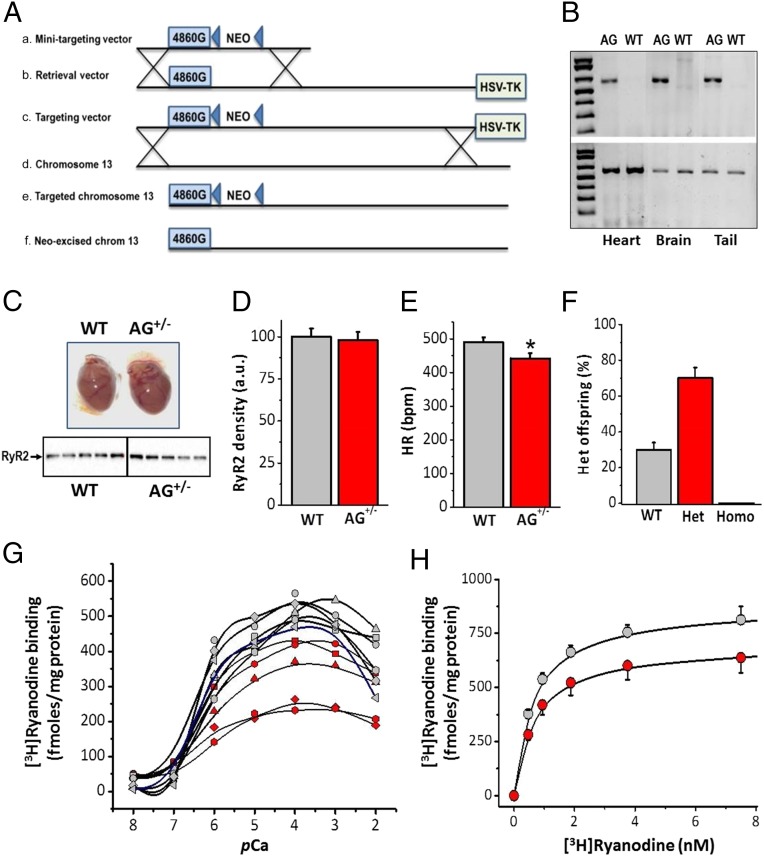
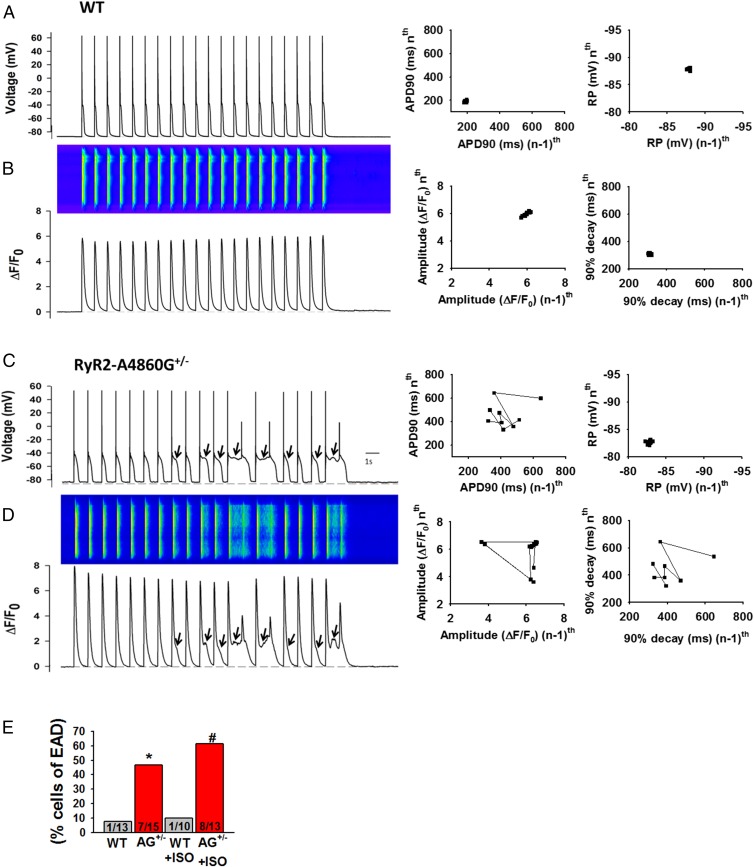
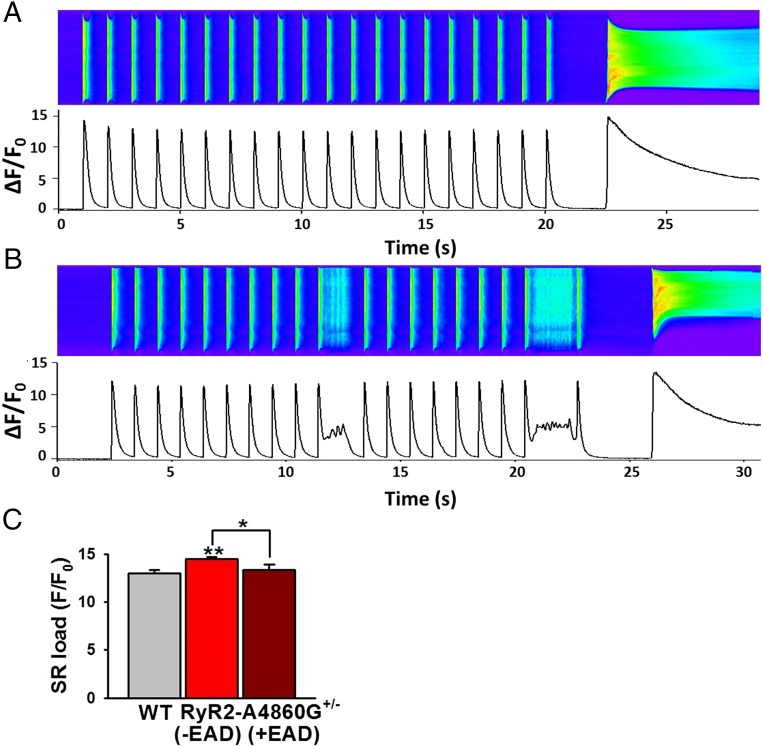
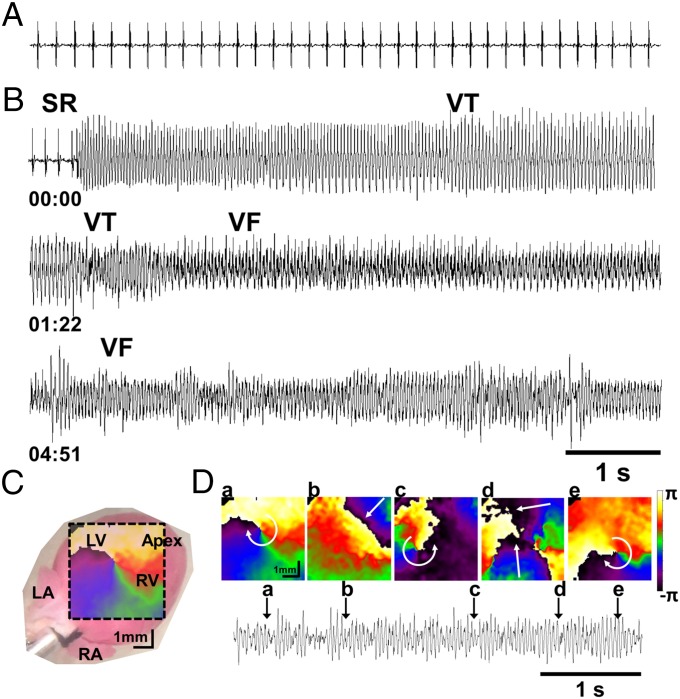
Current mechanisms of arrhythmogenesis in catecholaminergic polymorphic ventricular tachycardia (CPVT) require spontaneous Ca(2+) release via cardiac ryanodine receptor (RyR2) channels affected by gain-of-function mutations. Hence, hyperactive RyR2 channels eager to release Ca(2+) on their own appear as essential components of this arrhythmogenic scheme. This mechanism, therefore, appears inadequate to explain lethal arrhythmias in patients harboring RyR2 channels destabilized by loss-of-function mutations. We aimed to elucidate arrhythmia mechanisms in a RyR2-linked CPVT mutation (RyR2-A4860G) that depresses channel activity. Recombinant RyR2-A4860G protein was expressed equally as wild type (WT) RyR2, but channel activity was dramatically inhibited, as inferred by [(3)H]ryanodine binding and single channel recordings. Mice heterozygous for the RyR2-A4860G mutation (RyR2-A4860G(+/-)) exhibited basal bradycardia but no cardiac structural alterations; in contrast, no homozygotes were detected at birth, suggesting a lethal phenotype. Sympathetic stimulation elicited malignant arrhythmias in RyR2-A4860G(+/-) hearts, recapitulating the phenotype originally described in a human patient with the same mutation. In isoproterenol-stimulated ventricular myocytes, the RyR2-A4860G mutation decreased the peak of Ca(2+) release during systole, gradually overloading the sarcoplasmic reticulum with Ca(2+). The resultant Ca(2+) overload then randomly caused bursts of prolonged Ca(2+) release, activating electrogenic Na(+)-Ca(2+) exchanger activity and triggering early afterdepolarizations. The RyR2-A4860G mutation reveals novel pathways by which RyR2 channels engage sarcolemmal currents to produce life-threatening arrhythmias.
Keywords: CPVT; cardiac arrhythmias; heart; ryanodine receptor; sarcoplasmic reticulum.
Conflict of interest statement
The authors declare no conflict of interest.
Figures



Similar articles
-
Suppression of ryanodine receptor function prolongs Ca2+ release refractoriness and promotes cardiac alternans in intact hearts.Biochem J. 2016 Nov 1;473(21):3951-3964. doi: 10.1042/BCJ20160606. Epub 2016 Aug 31. Biochem J. 2016. PMID: 27582498 Free PMC article.
-
Na+-dependent SR Ca2+ overload induces arrhythmogenic events in mouse cardiomyocytes with a human CPVT mutation.Cardiovasc Res. 2010 Jul 1;87(1):50-9. doi: 10.1093/cvr/cvq007. Epub 2010 Jan 15. Cardiovasc Res. 2010. PMID: 20080988
-
Pathogenic mechanism of a catecholaminergic polymorphic ventricular tachycardia causing-mutation in cardiac calcium release channel RyR2.J Mol Cell Cardiol. 2018 Apr;117:26-35. doi: 10.1016/j.yjmcc.2018.02.014. Epub 2018 Mar 2. J Mol Cell Cardiol. 2018. PMID: 29477366
-
Sarcoplasmic Reticulum Ca2+ Dysregulation in the Pathophysiology of Inherited Arrhythmia: An Update.Biochem Pharmacol. 2022 Jun;200:115059. doi: 10.1016/j.bcp.2022.115059. Epub 2022 Apr 29. Biochem Pharmacol. 2022. PMID: 35490731 Review.
-
Catecholaminergic polymorphic ventricular tachycardia: recent mechanistic insights.Cardiovasc Res. 2005 Aug 15;67(3):379-87. doi: 10.1016/j.cardiores.2005.04.027. Cardiovasc Res. 2005. PMID: 15913575 Review.
Cited by
-
Dual ablation of the RyR2-Ser2808 and RyR2-Ser2814 sites increases propensity for pro-arrhythmic spontaneous Ca2+ releases.J Physiol. 2024 Oct;602(20):5179-5201. doi: 10.1113/JP286453. Epub 2024 Sep 24. J Physiol. 2024. PMID: 39316734
-
p38γ/δ activation alters cardiac electrical activity and predisposes to ventricular arrhythmia.Nat Cardiovasc Res. 2023 Dec;2(12):1204-1220. doi: 10.1038/s44161-023-00368-x. Epub 2023 Nov 27. Nat Cardiovasc Res. 2023. PMID: 39196141
-
Mitochondrial Calcium Regulation of Cardiac Metabolism in Health and Disease.Physiology (Bethesda). 2024 Sep 1;39(5):0. doi: 10.1152/physiol.00014.2024. Epub 2024 May 7. Physiology (Bethesda). 2024. PMID: 38713090 Review.
-
An inherited life-threatening arrhythmia model established by screening randomly mutagenized mice.Proc Natl Acad Sci U S A. 2024 Apr 23;121(17):e2218204121. doi: 10.1073/pnas.2218204121. Epub 2024 Apr 15. Proc Natl Acad Sci U S A. 2024. PMID: 38621141 Free PMC article.
-
Dissecting the roles of calcium cycling and its coupling with voltage in the genesis of early afterdepolarizations in cardiac myocyte models.PLoS Comput Biol. 2024 Feb 28;20(2):e1011930. doi: 10.1371/journal.pcbi.1011930. eCollection 2024 Feb. PLoS Comput Biol. 2024. PMID: 38416778 Free PMC article.
References
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
