

The treatment-naive microbiome in new-onset Crohn's disease
- PMID: 24629344
- PMCID: PMC4059512
- DOI: 10.1016/j.chom.2014.02.005
The treatment-naive microbiome in new-onset Crohn's disease
Abstract
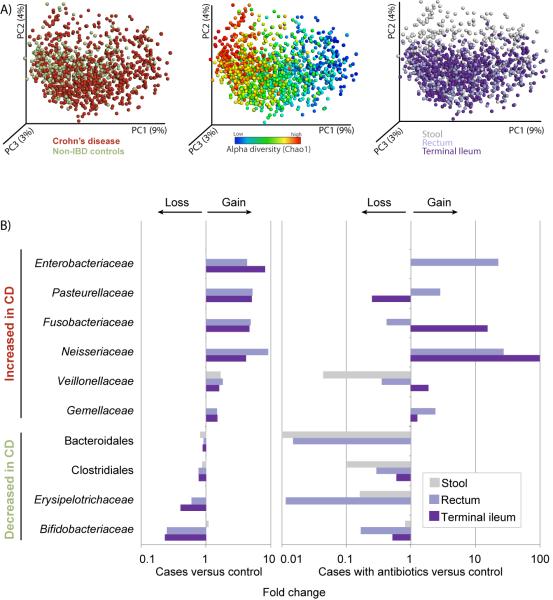
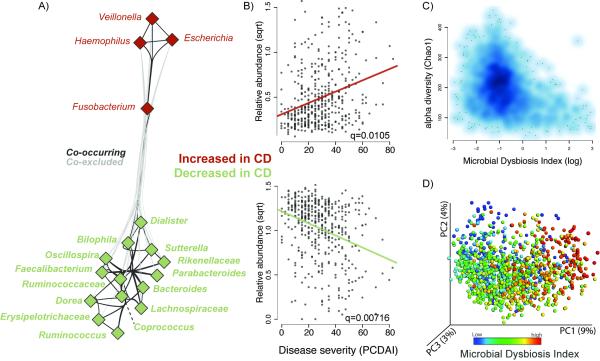
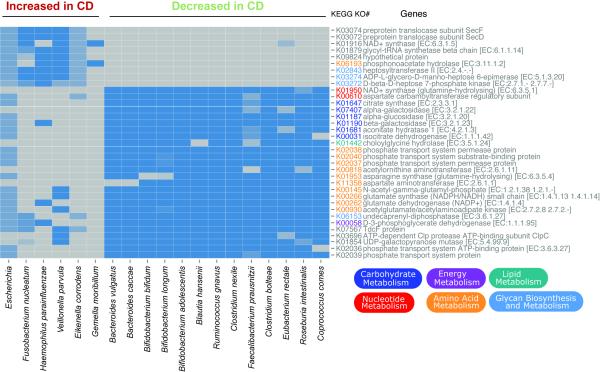
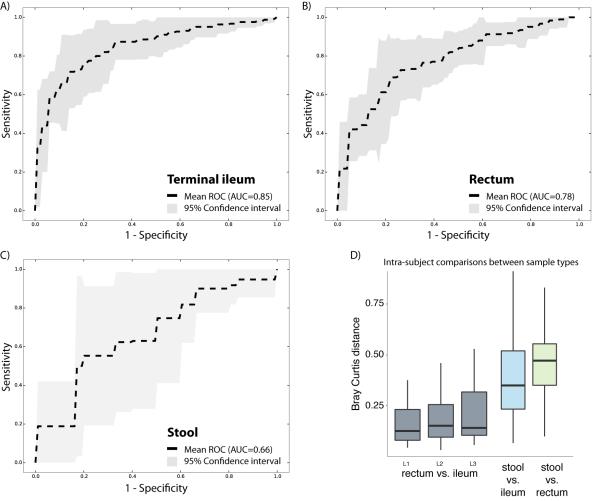
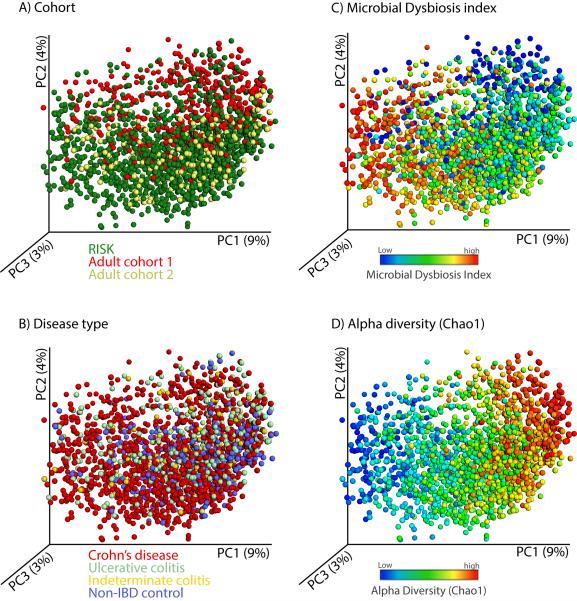
Inflammatory bowel diseases (IBDs), including Crohn's disease (CD), are genetically linked to host pathways that implicate an underlying role for aberrant immune responses to intestinal microbiota. However, patterns of gut microbiome dysbiosis in IBD patients are inconsistent among published studies. Using samples from multiple gastrointestinal locations collected prior to treatment in new-onset cases, we studied the microbiome in the largest pediatric CD cohort to date. An axis defined by an increased abundance in bacteria which include Enterobacteriaceae, Pasteurellacaea, Veillonellaceae, and Fusobacteriaceae, and decreased abundance in Erysipelotrichales, Bacteroidales, and Clostridiales, correlates strongly with disease status. Microbiome comparison between CD patients with and without antibiotic exposure indicates that antibiotic use amplifies the microbial dysbiosis associated with CD. Comparing the microbial signatures between the ileum, the rectum, and fecal samples indicates that at this early stage of disease, assessing the rectal mucosal-associated microbiome offers unique potential for convenient and early diagnosis of CD.
Copyright © 2014 Elsevier Inc. All rights reserved.
Figures
Comment in
-
Microbiome: bacterial imbalance in Crohn's disease.Nat Rev Microbiol. 2014 May;12(5):312. doi: 10.1038/nrmicro3255. Epub 2014 Mar 17. Nat Rev Microbiol. 2014. PMID: 24638106 No abstract available.
-
IBD. Understanding gut microbiota in new-onset Crohn's disease.Nat Rev Gastroenterol Hepatol. 2014 May;11(5):268. doi: 10.1038/nrgastro.2014.45. Epub 2014 Mar 25. Nat Rev Gastroenterol Hepatol. 2014. PMID: 24662277 No abstract available.
-
Gut microbiome in new-onset Crohn's disease.Gastroenterology. 2014 Oct;147(4):932-4. doi: 10.1053/j.gastro.2014.08.014. Epub 2014 Aug 23. Gastroenterology. 2014. PMID: 25152198 No abstract available.
Similar articles
-
Dysbiosis, inflammation, and response to treatment: a longitudinal study of pediatric subjects with newly diagnosed inflammatory bowel disease.Genome Med. 2016 Jul 13;8(1):75. doi: 10.1186/s13073-016-0331-y. Genome Med. 2016. PMID: 27412252 Free PMC article.
-
Gut microbiome in new-onset Crohn's disease.Gastroenterology. 2014 Oct;147(4):932-4. doi: 10.1053/j.gastro.2014.08.014. Epub 2014 Aug 23. Gastroenterology. 2014. PMID: 25152198 No abstract available.
-
IBD. Understanding gut microbiota in new-onset Crohn's disease.Nat Rev Gastroenterol Hepatol. 2014 May;11(5):268. doi: 10.1038/nrgastro.2014.45. Epub 2014 Mar 25. Nat Rev Gastroenterol Hepatol. 2014. PMID: 24662277 No abstract available.
-
Gut dysbiosis and paediatric Crohn's disease.J Infect. 2019 Jan;78(1):1-7. doi: 10.1016/j.jinf.2018.10.005. Epub 2018 Oct 16. J Infect. 2019. PMID: 30336176 Review.
-
The Microbiome in Crohn's Disease: Role in Pathogenesis and Role of Microbiome Replacement Therapies.Gastroenterol Clin North Am. 2017 Sep;46(3):481-492. doi: 10.1016/j.gtc.2017.05.004. Epub 2017 Jul 19. Gastroenterol Clin North Am. 2017. PMID: 28838410 Review.
Cited by
-
Recipient factors in faecal microbiota transplantation: one stool does not fit all.Nat Rev Gastroenterol Hepatol. 2021 Jul;18(7):503-513. doi: 10.1038/s41575-021-00441-5. Epub 2021 Apr 27. Nat Rev Gastroenterol Hepatol. 2021. PMID: 33907321 Review.
-
Low-FODMAP Diet for the Management of Irritable Bowel Syndrome in Remission of IBD.Nutrients. 2022 Oct 29;14(21):4562. doi: 10.3390/nu14214562. Nutrients. 2022. PMID: 36364824 Free PMC article.
-
The gut microbiota is associated with the small intestinal paracellular permeability and the development of the immune system in healthy children during the first two years of life.J Transl Med. 2021 Apr 28;19(1):177. doi: 10.1186/s12967-021-02839-w. J Transl Med. 2021. PMID: 33910577 Free PMC article.
-
Alterations in Gut Microbial Communities Across Anatomical Locations in Inflammatory Bowel Diseases.Front Nutr. 2021 Feb 26;8:615064. doi: 10.3389/fnut.2021.615064. eCollection 2021. Front Nutr. 2021. PMID: 33718417 Free PMC article.
-
Snow microbiome functional analyses reveal novel aspects of microbial metabolism of complex organic compounds.Microbiologyopen. 2020 Sep;9(9):e1100. doi: 10.1002/mbo3.1100. Epub 2020 Aug 6. Microbiologyopen. 2020. PMID: 32762019 Free PMC article.
References
-
- Chao A, Chazdon RL, Colwell RK, Shen TJ. Abundance-based similarity indices and their estimation when there are unseen species in samples. Biometrics. 2006;62:361–371. - PubMed
-
- Chavan RS, Pannaraj PS, Luna RA, Szabo S, Adesina A, Versalovic J, Krance RA, Kennedy-Nasser AA. Significant morbidity and mortality attributable to rothia mucilaginosa infections in children with hematological malignancies or following hematopoietic stem cell transplantation. Pediatr Hematol Oncol. 2013;30:445–454. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases