

Development of personalized tumor biomarkers using massively parallel sequencing
- PMID: 20371490
- PMCID: PMC2858564
- DOI: 10.1126/scitranslmed.3000702
Development of personalized tumor biomarkers using massively parallel sequencing
Abstract
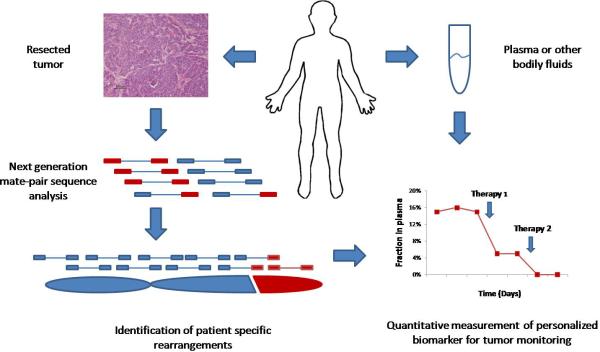
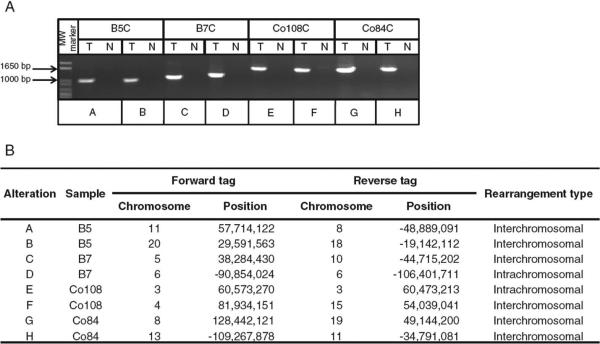
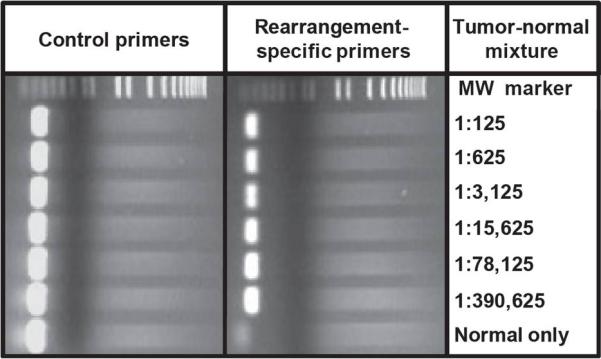
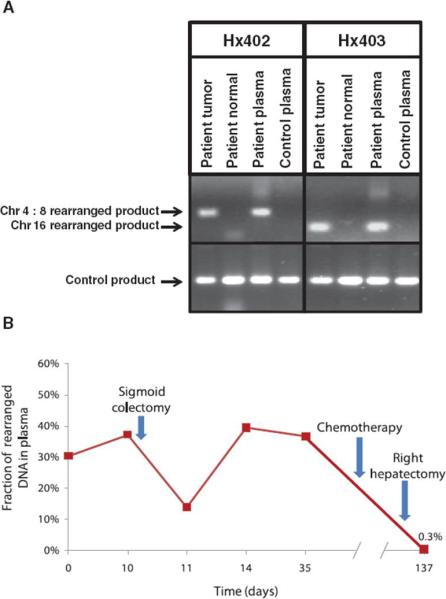
Clinical management of human cancer is dependent on the accurate monitoring of residual and recurrent tumors. The evaluation of patient-specific translocations in leukemias and lymphomas has revolutionized diagnostics for these diseases. We have developed a method, called personalized analysis of rearranged ends (PARE), which can identify translocations in solid tumors. Analysis of four colorectal and two breast cancers with massively parallel sequencing revealed an average of nine rearranged sequences (range, 4 to 15) per tumor. Polymerase chain reaction with primers spanning the breakpoints was able to detect mutant DNA molecules present at levels lower than 0.001% and readily identified mutated circulating DNA in patient plasma samples. This approach provides an exquisitely sensitive and broadly applicable approach for the development of personalized biomarkers to enhance the clinical management of cancer patients.
Figures
Comment in
-
Cancer sequencing gets a little more personal.Sci Transl Med. 2010 Feb 24;2(20):20ps8. doi: 10.1126/scitranslmed.3000929. Sci Transl Med. 2010. PMID: 20371489
Similar articles
-
Identification and Use of Personalized Genomic Markers for Monitoring Circulating Tumor DNA.Methods Mol Biol. 2018;1768:303-322. doi: 10.1007/978-1-4939-7778-9_17. Methods Mol Biol. 2018. PMID: 29717450
-
Digital PCR of Genomic Rearrangements for Monitoring Circulating Tumour DNA.Adv Exp Med Biol. 2016;924:139-146. doi: 10.1007/978-3-319-42044-8_27. Adv Exp Med Biol. 2016. PMID: 27753035
-
Simultaneous evaluation of T- and B-cell clonality, t(11;14) and t(14;18), in a single reaction by a four-color multiplex polymerase chain reaction assay and automated high-resolution fragment analysis: a method for the rapid molecular diagnosis of lymphoproliferative disorders applicable to fresh frozen and formalin-fixed, paraffin-embedded tissues, blood, and bone marrow aspirates.Am J Pathol. 2001 Dec;159(6):2031-43. doi: 10.1016/S0002-9440(10)63055-6. Am J Pathol. 2001. PMID: 11733354 Free PMC article.
-
Using circulating cell-free DNA to monitor personalized cancer therapy.Crit Rev Clin Lab Sci. 2017 May;54(3):205-218. doi: 10.1080/10408363.2017.1299683. Epub 2017 Apr 10. Crit Rev Clin Lab Sci. 2017. PMID: 28393575 Review.
-
Comprehensive next-generation cancer genome sequencing in the era of targeted therapy and personalized oncology.Biomark Med. 2011 Jun;5(3):293-305. doi: 10.2217/bmm.11.37. Biomark Med. 2011. PMID: 21657839 Review.
Cited by
-
Establishment of tumor-specific copy number alterations from plasma DNA of patients with cancer.Int J Cancer. 2013 Jul 15;133(2):346-56. doi: 10.1002/ijc.28030. Epub 2013 Feb 13. Int J Cancer. 2013. PMID: 23319339 Free PMC article.
-
Transflip mutations produce deletions in pancreatic cancer.Genes Chromosomes Cancer. 2015 Aug;54(8):472-481. doi: 10.1002/gcc.22258. Epub 2015 May 29. Genes Chromosomes Cancer. 2015. PMID: 26031834 Free PMC article.
-
Circulating biomarkers to monitor cancer progression and treatment.Comput Struct Biotechnol J. 2016 Jun 1;14:211-22. doi: 10.1016/j.csbj.2016.05.004. eCollection 2016. Comput Struct Biotechnol J. 2016. PMID: 27358717 Free PMC article. Review.
-
Systems cancer medicine: towards realization of predictive, preventive, personalized and participatory (P4) medicine.J Intern Med. 2012 Feb;271(2):111-21. doi: 10.1111/j.1365-2796.2011.02498.x. J Intern Med. 2012. PMID: 22142401 Free PMC article. Review.
-
Identification of medium-sized genomic deletions with low coverage, mate-paired restricted tags.BMC Genomics. 2013 Jan 24;14:51. doi: 10.1186/1471-2164-14-51. BMC Genomics. 2013. PMID: 23347462 Free PMC article.
References
-
- Lengauer C, Kinzler KW, Vogelstein B. Genetic instabilities in human cancers. Nature. 1998;396:643–649. - PubMed
-
- Mitelman F, Johansson B, Mertens F. The impact of translocations and gene fusions on cancer causation. Nat. Rev. Cancer. 2007;7:233–245. - PubMed
-
- Pinkel D, Segraves R, Sudar D, Clark S, Poole I, Kowbel D, Collins C, Kuo WL, Chen C, Zhai Y, Dairkee SH, Ljung BM, Gray JW, Albertson DG. High resolution analysis of DNA copy number variation using comparative genomic hybridization to microarrays. Nat. Genet. 1998;20:207–211. - PubMed
-
- Lucito R, Healy J, Alexander J, Reiner A, Esposito D, Chi M, Rodgers L, Brady A, Sebat J, Troge J, West JA, Rostan S, Nguyen KC, Powers S, Ye KQ, Olshen A, Venkatraman E, Norton L, Wigler M. Representational oligonucleotide microarray analysis: A high-resolution method to detect genome copy number variation. Genome Res. 2003;13:2291–2305. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
