

The mitochondrial uncoupling protein-2 promotes chemoresistance in cancer cells
- PMID: 18413749
- PMCID: PMC2386271
- DOI: 10.1158/0008-5472.CAN-08-0053
The mitochondrial uncoupling protein-2 promotes chemoresistance in cancer cells
Abstract
Cancer cells acquire drug resistance as a result of selection pressure dictated by unfavorable microenvironments. This survival process is facilitated through efficient control of oxidative stress originating from mitochondria that typically initiates programmed cell death. We show this critical adaptive response in cancer cells to be linked to uncoupling protein-2 (UCP2), a mitochondrial suppressor of reactive oxygen species (ROS). UCP2 is present in drug-resistant lines of various cancer cells and in human colon cancer. Overexpression of UCP2 in HCT116 human colon cancer cells inhibits ROS accumulation and apoptosis after exposure to chemotherapeutic agents. Tumor xenografts of UCP2-overexpressing HCT116 cells retain growth in nude mice receiving chemotherapy. Augmented cancer cell survival is accompanied by altered NH(2)-terminal phosphorylation of the pivotal tumor suppressor p53 and induction of the glycolytic phenotype (Warburg effect). These findings link UCP2 with molecular mechanisms of chemoresistance. Targeting UCP2 may be considered a novel treatment strategy for cancer.
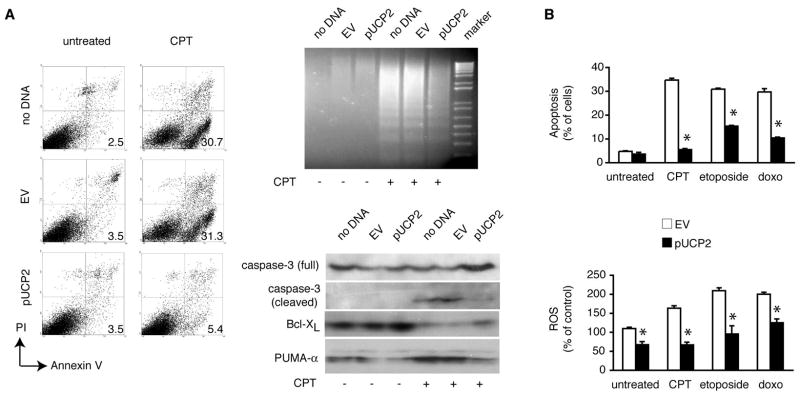
Figures




Similar articles
-
Increased activity of mitochondrial uncoupling protein 2 improves stress resistance in cultured endothelial cells exposed in vitro to high glucose levels.Am J Physiol Heart Circ Physiol. 2015 Jul 1;309(1):H147-56. doi: 10.1152/ajpheart.00759.2014. Epub 2015 Apr 24. Am J Physiol Heart Circ Physiol. 2015. PMID: 25910810
-
Uncoupling protein 2 mediates resistance to gemcitabine-induced apoptosis in hepatocellular carcinoma cell lines.Biosci Rep. 2015 Jun 16;35(4):e00231. doi: 10.1042/BSR20150116. Biosci Rep. 2015. PMID: 26181366 Free PMC article.
-
UCP2-related mitochondrial pathway participates in oroxylin A-induced apoptosis in human colon cancer cells.J Cell Physiol. 2015 May;230(5):1054-63. doi: 10.1002/jcp.24833. J Cell Physiol. 2015. PMID: 25251374
-
Overexpression of uncoupling protein-2 in cancer: metabolic and heat changes, inhibition and effects on drug resistance.Inflammopharmacology. 2015 Dec;23(6):365-9. doi: 10.1007/s10787-015-0250-3. Epub 2015 Nov 5. Inflammopharmacology. 2015. PMID: 26542482 Review.
-
Mitochondrial recoupling: a novel therapeutic strategy for cancer?Br J Cancer. 2011 Aug 9;105(4):469-74. doi: 10.1038/bjc.2011.245. Epub 2011 Jun 28. Br J Cancer. 2011. PMID: 21712825 Free PMC article. Review.
Cited by
-
Mitochondrial Uncoupling Protein-2 Ameliorates Ischemic Stroke by Inhibiting Ferroptosis-Induced Brain Injury and Neuroinflammation.Mol Neurobiol. 2024 Jun 14. doi: 10.1007/s12035-024-04288-0. Online ahead of print. Mol Neurobiol. 2024. PMID: 38874704
-
Uncoupling Protein 2 Alleviates Myocardial Ischemia/Reperfusion Injury by Inhibiting Cardiomyocyte Ferroptosis.J Vasc Res. 2024;61(3):109-121. doi: 10.1159/000537925. Epub 2024 Apr 12. J Vasc Res. 2024. PMID: 38615660 Free PMC article.
-
UCP2 and pancreatic cancer: conscious uncoupling for therapeutic effect.Cancer Metastasis Rev. 2024 Jun;43(2):777-794. doi: 10.1007/s10555-023-10157-4. Epub 2024 Jan 9. Cancer Metastasis Rev. 2024. PMID: 38194152 Free PMC article. Review.
-
Mitochondrial transfer in hematological malignancies.Biomark Res. 2023 Oct 5;11(1):89. doi: 10.1186/s40364-023-00529-x. Biomark Res. 2023. PMID: 37798791 Free PMC article. Review.
-
CircUCP2 promotes the tumor progression of non-small cell lung cancer through the miR-149/UCP2 pathway.Oncol Res. 2023 Sep 15;31(6):929-936. doi: 10.32604/or.2023.030611. eCollection 2023. Oncol Res. 2023. PMID: 37744277 Free PMC article.
References
-
- Hussain SP, Hofseth LJ, Harris CC. Radical causes of cancer. Nat Rev Cancer. 2003;3:276–85. - PubMed
-
- Martindale JL, Holbrook NJ. Cellular response to oxidative stress: signaling for suicide and survival. J Cell Physiol. 2002;192:1–15. - PubMed
-
- Schumacker PT. Reactive oxygen species in cancer cells: live by the sword, die by the sword. Cancer Cell. 2006;10:175–6. - PubMed
-
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. - PubMed
-
- Halliwell B. Oxidative stress and cancer: have we moved forward? Biochem J. 2007;401:1–11. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
