

FLAME, a novel fuzzy clustering method for the analysis of DNA microarray data
- PMID: 17204155
- PMCID: PMC1774579
- DOI: 10.1186/1471-2105-8-3
FLAME, a novel fuzzy clustering method for the analysis of DNA microarray data
Abstract
Background: Data clustering analysis has been extensively applied to extract information from gene expression profiles obtained with DNA microarrays. To this aim, existing clustering approaches, mainly developed in computer science, have been adapted to microarray data analysis. However, previous studies revealed that microarray datasets have very diverse structures, some of which may not be correctly captured by current clustering methods. We therefore approached the problem from a new starting point, and developed a clustering algorithm designed to capture dataset-specific structures at the beginning of the process.
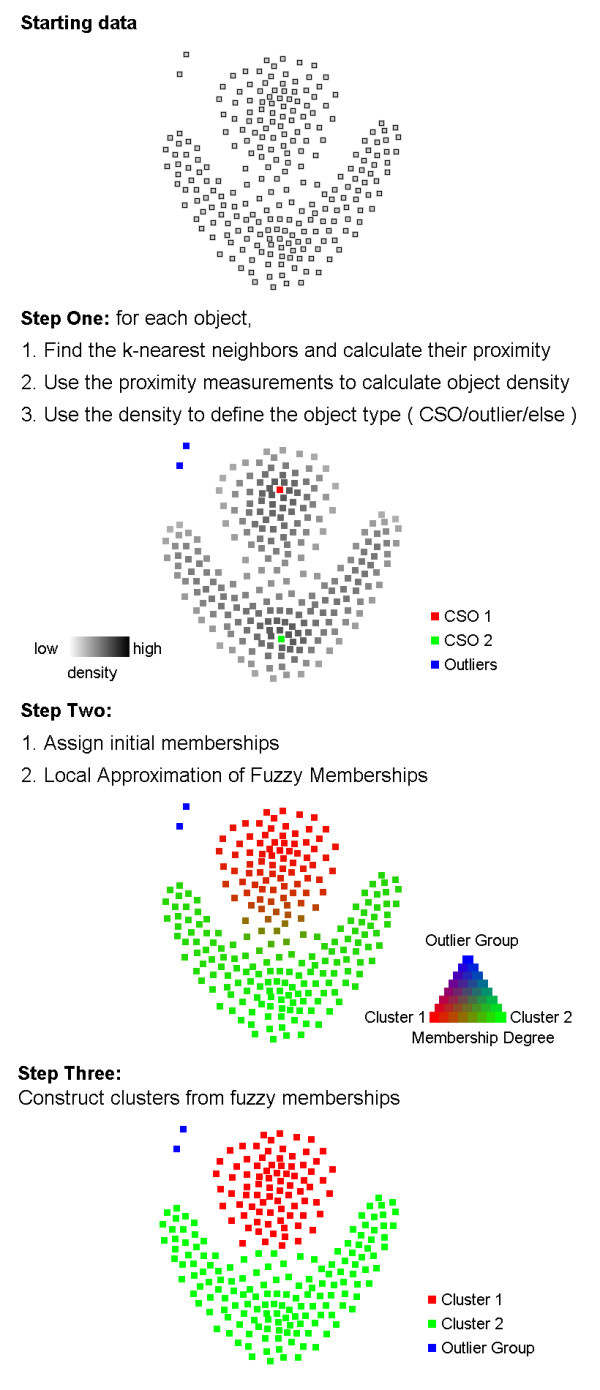
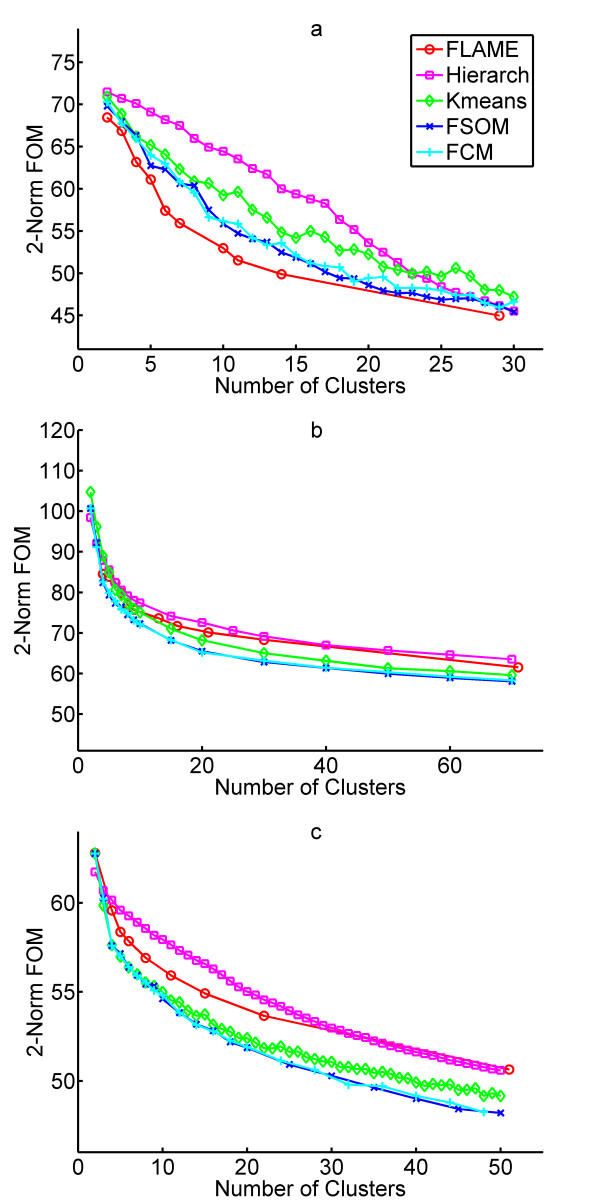
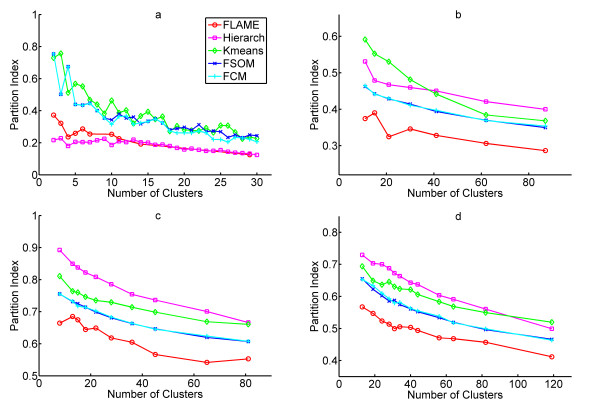
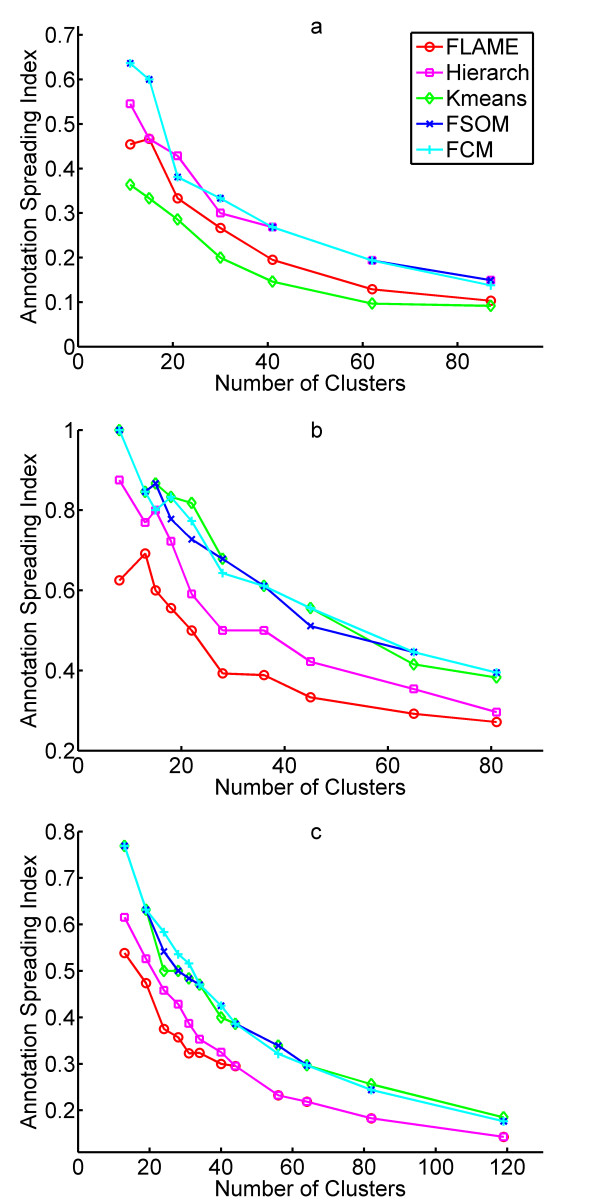
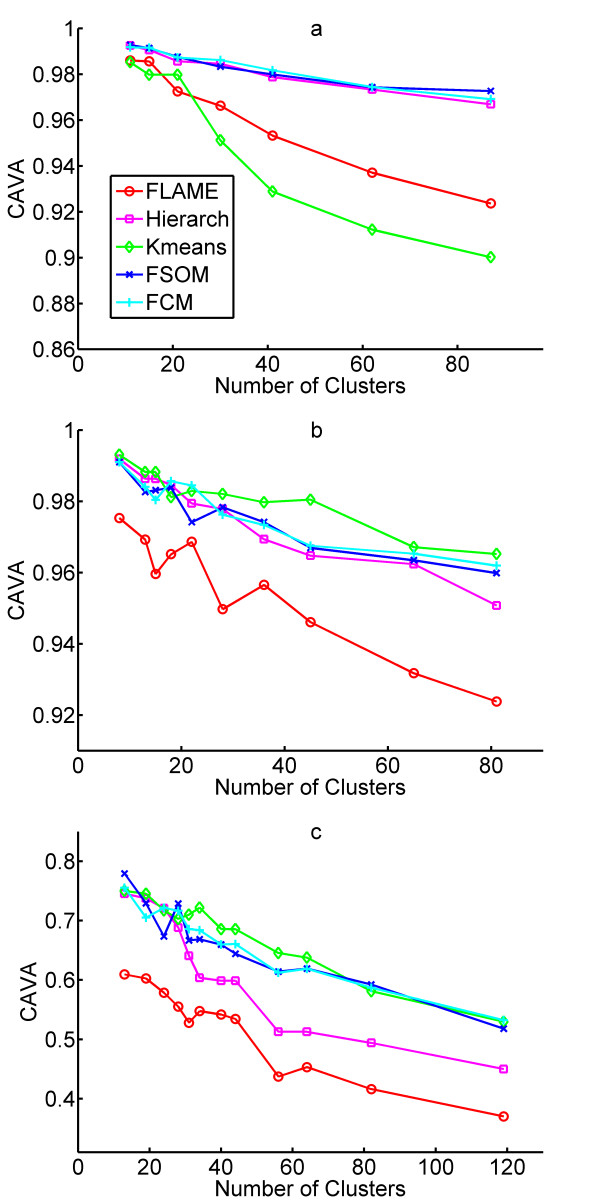
Results: The clustering algorithm is named Fuzzy clustering by Local Approximation of MEmbership (FLAME). Distinctive elements of FLAME are: (i) definition of the neighborhood of each object (gene or sample) and identification of objects with "archetypal" features named Cluster Supporting Objects, around which to construct the clusters; (ii) assignment to each object of a fuzzy membership vector approximated from the memberships of its neighboring objects, by an iterative converging process in which membership spreads from the Cluster Supporting Objects through their neighbors. Comparative analysis with K-means, hierarchical, fuzzy C-means and fuzzy self-organizing maps (SOM) showed that data partitions generated by FLAME are not superimposable to those of other methods and, although different types of datasets are better partitioned by different algorithms, FLAME displays the best overall performance. FLAME is implemented, together with all the above-mentioned algorithms, in a C++ software with graphical interface for Linux and Windows, capable of handling very large datasets, named Gene Expression Data Analysis Studio (GEDAS), freely available under GNU General Public License.
Conclusion: The FLAME algorithm has intrinsic advantages, such as the ability to capture non-linear relationships and non-globular clusters, the automated definition of the number of clusters, and the identification of cluster outliers, i.e. genes that are not assigned to any cluster. As a result, clusters are more internally homogeneous and more diverse from each other, and provide better partitioning of biological functions. The clustering algorithm can be easily extended to applications different from gene expression analysis.
Figures
Similar articles
-
Detecting clusters of different geometrical shapes in microarray gene expression data.Bioinformatics. 2005 May 1;21(9):1927-34. doi: 10.1093/bioinformatics/bti251. Epub 2005 Jan 12. Bioinformatics. 2005. PMID: 15647300
-
Visualization methods for statistical analysis of microarray clusters.BMC Bioinformatics. 2005 May 12;6:115. doi: 10.1186/1471-2105-6-115. BMC Bioinformatics. 2005. PMID: 15890080 Free PMC article.
-
A new validity measure for a correlation-based fuzzy c-means clustering algorithm.Annu Int Conf IEEE Eng Med Biol Soc. 2009;2009:3865-8. doi: 10.1109/IEMBS.2009.5332582. Annu Int Conf IEEE Eng Med Biol Soc. 2009. PMID: 19963601 Free PMC article.
-
Clustering approaches to identifying gene expression patterns from DNA microarray data.Mol Cells. 2008 Apr 30;25(2):279-88. Epub 2008 Mar 31. Mol Cells. 2008. PMID: 18414008 Review.
-
Inference from clustering with application to gene-expression microarrays.J Comput Biol. 2002;9(1):105-26. doi: 10.1089/10665270252833217. J Comput Biol. 2002. PMID: 11911797 Review.
Cited by
-
Gene expression profiling of HGF/Met activation in neonatal mouse heart.Transgenic Res. 2013 Jun;22(3):579-93. doi: 10.1007/s11248-012-9667-2. Epub 2012 Dec 6. Transgenic Res. 2013. PMID: 23224784
-
Comparative analysis of missing value imputation methods to improve clustering and interpretation of microarray experiments.BMC Genomics. 2010 Jan 7;11:15. doi: 10.1186/1471-2164-11-15. BMC Genomics. 2010. PMID: 20056002 Free PMC article.
-
Clusterdv: a simple density-based clustering method that is robust, general and automatic.Bioinformatics. 2019 Jun 1;35(12):2125-2132. doi: 10.1093/bioinformatics/bty932. Bioinformatics. 2019. PMID: 30407500 Free PMC article.
-
The thioxotriazole copper(II) complex A0 induces endoplasmic reticulum stress and paraptotic death in human cancer cells.J Biol Chem. 2009 Sep 4;284(36):24306-19. doi: 10.1074/jbc.M109.026583. Epub 2009 Jun 26. J Biol Chem. 2009. PMID: 19561079 Free PMC article.
-
TNF-α promotes invasive growth through the MET signaling pathway.Mol Oncol. 2015 Feb;9(2):377-88. doi: 10.1016/j.molonc.2014.09.002. Epub 2014 Sep 26. Mol Oncol. 2015. PMID: 25306394 Free PMC article.
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
